Related Research Articles

Epilepsy is a group of non-communicable neurological disorders characterized by recurrent epileptic seizures. An epileptic seizure is the clinical manifestation of an abnormal, excessive, and synchronized electrical discharge in the neurons. The occurrence of two or more unprovoked seizures defines epilepsy. The occurrence of just one seizure may warrant the definition in a more clinical usage where recurrence may be able to be prejudged. Epileptic seizures can vary from brief and nearly undetectable periods to long periods of vigorous shaking due to abnormal electrical activity in the brain. These episodes can result in physical injuries, either directly such as broken bones or through causing accidents. In epilepsy, seizures tend to recur and may have no detectable underlying cause. Isolated seizures that are provoked by a specific cause such as poisoning are not deemed to represent epilepsy. People with epilepsy may be treated differently in various areas of the world and experience varying degrees of social stigma due to the alarming nature of their symptoms.
A seizure is a period of symptoms due to abnormally excessive or synchronous neuronal activity in the brain. Outward effects vary from uncontrolled shaking movements involving much of the body with loss of consciousness, to shaking movements involving only part of the body with variable levels of consciousness, to a subtle momentary loss of awareness. These episodes usually last less than two minutes and it takes some time to return to normal. Loss of bladder control may occur.
Anticonvulsants are a diverse group of pharmacological agents used in the treatment of epileptic seizures. Anticonvulsants are also increasingly being used in the treatment of bipolar disorder and borderline personality disorder, since many seem to act as mood stabilizers, and for the treatment of neuropathic pain. Anticonvulsants suppress the excessive rapid firing of neurons during seizures. Anticonvulsants also prevent the spread of the seizure within the brain.

Long QT syndrome (LQTS) is a condition affecting repolarization (relaxing) of the heart after a heartbeat, giving rise to an abnormally lengthy QT interval. It results in an increased risk of an irregular heartbeat which can result in fainting, drowning, seizures, or sudden death. These episodes can be triggered by exercise or stress. Some rare forms of LQTS are associated with other symptoms and signs including deafness and periods of muscle weakness.
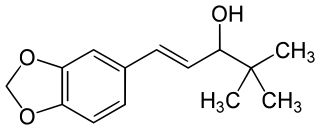
Stiripentol, sold under the brand name Diacomit, is an anticonvulsant medication used for the treatment of Dravet syndrome - a serious genetic brain disorder.
Sodium channels are integral membrane proteins that form ion channels, conducting sodium ions (Na+) through a cell's membrane. They belong to the superfamily of cation channels.
Sudden unexpected death in epilepsy (SUDEP) is a fatal complication of epilepsy. It is defined as the sudden and unexpected, non-traumatic and non-drowning death of a person with epilepsy, without a toxicological or anatomical cause of death detected during the post-mortem examination.
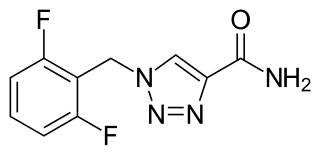
Rufinamide is an anticonvulsant medication. It is used in combination with other medication and therapy to treat Lennox–Gastaut syndrome and various other seizure disorders. Rufinamide, a triazole derivative, was developed in 2004 by Novartis Pharma, AG, and is manufactured by Eisai.
Generalized epilepsy with febrile seizures plus (GEFS+) is a syndromic autosomal dominant disorder where affected individuals can exhibit numerous epilepsy phenotypes. GEFS+ can persist beyond early childhood. GEFS+ is also now believed to encompass three other epilepsy disorders: severe myoclonic epilepsy of infancy (SMEI), which is also known as Dravet's syndrome, borderline SMEI (SMEB), and intractable epilepsy of childhood (IEC). There are at least six types of GEFS+, delineated by their causative gene. Known causative gene mutations are in the sodium channel α subunit genes SCN1A, an associated β subunit SCN1B, and in a GABAA receptor γ subunit gene, in GABRG2 and there is another gene related with calcium channel the PCDH19 which is also known as Epilepsy Female with Mental Retardation. Penetrance for this disorder is estimated at 60%.
Dravet syndrome (DS), previously known as severe myoclonic epilepsy of infancy (SMEI), is an autosomal dominant genetic disorder which causes a catastrophic form of epilepsy, with prolonged seizures that are often triggered by hot temperatures or fever. It is very difficult to treat with anticonvulsant medications. It often begins before one year of age, with six months being the age that seizures, characterized by prolonged convulsions and triggered by fever, usually begin.

Sodium channel protein type 5 subunit alpha, also known as NaV1.5 is an integral membrane protein and tetrodotoxin-resistant voltage-gated sodium channel subunit. NaV1.5 is found primarily in cardiac muscle, where it mediates the fast influx of Na+-ions (INa) across the cell membrane, resulting in the fast depolarization phase of the cardiac action potential. As such, it plays a major role in impulse propagation through the heart. A vast number of cardiac diseases is associated with mutations in NaV1.5 (see paragraph genetics). SCN5A is the gene that encodes the cardiac sodium channel NaV1.5.
Paralytic is a gene in the fruit fly, Drosophila melanogaster, which encodes a voltage gated sodium channel within D. melanogaster neurons. This gene is essential for locomotive activity in the fly. There are 9 different para alleles, composed of a minimum of 26 exons within over 78kb of genomic DNA. The para gene undergoes alternative splicing to produce subtypes of the channel protein. Flies with mutant forms of paralytic are used in fly models of seizures, since seizures can be easily induced in these flies.

CDKL5 is a gene that provides instructions for making a protein called cyclin-dependent kinase-like 5 also known as serine/threonine kinase 9 (STK9) that is essential for normal brain development. Mutations in the gene can cause deficiencies in the protein. The gene regulates neuronal morphology through cytoplasmic signaling and controlling gene expression. The CDKL5 protein acts as a kinase, which is an enzyme that changes the activity of other proteins by adding a cluster of oxygen and phosphorus atoms at specific positions. Researchers are currently working to determine which proteins are targeted by the CDKL5 protein.

Sodium channel protein type 1 subunit alpha (SCN1A), is a protein which in humans is encoded by the SCN1A gene.
Spike-and-wave is a pattern of the electroencephalogram (EEG) typically observed during epileptic seizures. A spike-and-wave discharge is a regular, symmetrical, generalized EEG pattern seen particularly during absence epilepsy, also known as ‘petit mal’ epilepsy. The basic mechanisms underlying these patterns are complex and involve part of the cerebral cortex, the thalamocortical network, and intrinsic neuronal mechanisms.

Potassium voltage-gated channel subfamily C member 1 is a protein that in humans is encoded by the KCNC1 gene.
GABA transporters (Gamma-Aminobutyric acid transporters) belong to the family of neurotransmitters known as sodium symporters, also known as solute carrier 6 (SLC6). These are large family of neurotransmitter which are Na+ concentration dependent. They are found in various regions of the brain in different cell types, such as neurons and astrocytes.
Epilepsy-intellectual disability in females also known as PCDH19 gene-related epilepsy or epileptic encephalopathy, early infantile, 9 (EIEE9), is a rare type of epilepsy that affects predominately females and is characterized by clusters of brief seizures, which start in infancy or early childhood, and is occasionally accompanied by varying degrees of cognitive impairment. The striking pattern of onset seizures at a young age, genetic testing and laboratory results, potential developmental delays or developmental regression and associated disorders, eases diagnosis.
Gene therapy is being studied for some forms of epilepsy. It relies on viral or non-viral vectors to deliver DNA or RNA to target brain areas where seizures arise, in order to prevent the development of epilepsy or to reduce the frequency and/or severity of seizures. Gene therapy has delivered promising results in early stage clinical trials for other neurological disorders such as Parkinson's disease, raising the hope that it will become a treatment for intractable epilepsy.

Charles Antzelevitch is an American cardiovascular research scientist in the fields of cardiac electrophysiology and cardiac arrhythmia syndromes.
References
- ↑ "Lori L. Isom, Ph.D. | Pharmacology | Michigan Medicine | University of Michigan". 9 December 2014.
- ↑ "Saving Lives with Basic Science | Medicine at Michigan". 21 November 2018.
- ↑ "Lori L. Isom, Ph.D. | Pharmacology | Michigan Medicine | University of Michigan". 9 December 2014.
- ↑ http://medicindeatmichigan.org/faculty/2018/fall/saving-lives-basic-science%5B%5D
- ↑ "Lori L. Isom, Ph.D. | Pharmacology | Michigan Medicine | University of Michigan". 9 December 2014.
- ↑ "National Academy of Medicine Elects 100 New Members". National Academy of Medicine. 18 October 2021. Retrieved 22 October 2021.
| Authority control databases: Academics |
|---|