This condition is inherited via autosomal dominant manner.
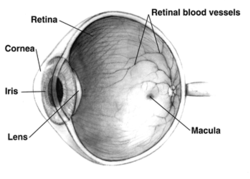
Macular hypoplasia (or foveal hypoplasia) is a rare medical condition involving the underdevelopment of the macula,[1] a small area on the retina (the eye's internal surface) responsible for seeing in detail and sensing light.[2] Macular hypoplasia is often associated with albinism.[1]
When the foveal area of the eye is compromised, visual clarity and color perception are reduced.[2] Diagnosing is done by an ophthalmologist.[2] The foveal area of the eye is located in the back of the eyeball. It is placed in front of the optic nerve and is responsible for light sensory and visual perceptiveness.[3][4]
This section is empty. You can help by adding to it. (November 2025)
Causes
Macular hypoplasia occurs the most in people that have a diagnosis of albinism.[7] There are four gene mutations that occur in albinism and are linked to macular hypoplasia.[7] The four mutations can occur on the phenotypes of FH, PAX6, SLC38A8, and AHR.[7] The most common gene mutation is the FH phenotype and has a 67.5% correlation rate to macular hypoplasia.[7]
The disorder can occur through two distinct genetic abnormalities.[8] The difference among mutated genes results in a difference in phenotypic display of macular hypoplasia.[8] In phenotype FVH1, there is a mutation of the PAX6 gene.[8] FVH1 occurs through autosomal dominant inheritance.[8] The mutation is passed down to the recipient from the mother or father.[8] FVH1 type of macular hypoplasia coincides with cataracts in the eyes.[8] In phenotype, FVH1 is caused by a SLC38A8 gene mutation. FVH2 occurs by autosomal recessive inheritance.[8] Both parents pass the mutated gene to the child.[8] Macular hypoplasia prevails due to improper placement of the optic nerve.[8]
Diagnosis
A lack of foveal pigmentation or circumfoveal light reflex is a common finding of macular hypoplasia; however, diagnosis is challenging for those that have a darker pigmentation of the skin, hair, and iris.[9] Originally, findings of nystagmus, or involuntary movement, and lack of blood flow to the retina using fluorescein angiography (FA) were used to detect macular hypoplasia.[9][10] FA uses light to look at the retina and blood vessel development in the eye using a dye.[9]
Today, a newer technology, optical coherence tomography (OCT) is used to detect foveal hypoplasia and does not require a dye.[9] OCT allows professionals to see the structures in the eye, usually the thickness of the retina and optic nerve.[11] This is a noninvasive procedure where patients rest their chin and focus on a green light within the machine.[11]Eye dryness and fatigue are the limited risks associated with this scan.[11]
Currently, there is no specific pharmacotherapy that prevents or reserves macular hypoplasia; however, reading glasses or other vision devices can be used to enhance the quality of life for individuals.[12]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.