
A lipoma is a benign tumor made of fat tissue. They are generally soft to the touch, movable, and painless. They usually occur just under the skin, but occasionally may be deeper. Most are less than 5 cm (2.0 in) in size. Common locations include upper back, shoulders, and abdomen. It is possible to have a number of lipomas.

Cutaneous squamous-cell carcinoma (cSCC), or squamous-cell carcinoma of the skin, also known as squamous-cell skin cancer, is, with basal-cell carcinoma and melanoma, one of the three principal types of skin cancer. cSCC typically presents as a hard lump with a scaly top layer, but it may instead form an ulcer. Onset often occurs over a period of months. Cutaneous squamous-cell carcinoma is more likely to spread to distant areas than basal cell cancer. When confined to the outermost layer of the skin, a pre-invasive, or in situ, form of cSCC is known as Bowen's disease.

Acral lentiginous melanoma is an aggressive type of skin cancer. Melanoma is a group of serious skin cancers that arise from pigment cells (melanocytes); acral lentiginous melanoma is a kind of lentiginous skin melanoma. Acral lentiginous melanoma is the most common subtype in people with darker skins and is rare in people with lighter skin types. It is not caused by exposure to sunlight or UV radiation, and wearing sunscreen does not protect against it. Acral lentiginous melanoma is commonly found on the palms, soles, under the nails, and in the oral mucosa. It occurs on non-hair-bearing surfaces of the body, which have not necessarily been exposed to sunlight. It is also found on mucous membranes.

Dermatofibrosarcoma protuberans (DFSP) is a rare locally aggressive malignant cutaneous soft-tissue sarcoma. DFSP develops in the connective tissue cells in the middle layer of the skin (dermis). Estimates of the overall occurrence of DFSP in the United States are 0.8 to 4.5 cases per million persons per year. In the United States, DFSP accounts for between 1 and 6 percent of all soft-tissue sarcomas and 18 percent of all cutaneous soft-tissue sarcomas. In the Surveillance, Epidemiology and End Results (SEER) tumor registry from 1992 through 2004, DFSP was second only to Kaposi sarcoma.

Invasive carcinoma of no special type, invasive breast carcinoma of no special type (IBC-NST), invasive ductal carcinoma (IDC), infiltrating ductal carcinoma (IDC) or invasive ductal carcinoma, not otherwise specified (NOS) is a disease. For international audiences this article will use "invasive carcinoma NST" because it is the preferred term of the World Health Organization (WHO).

Hemangioendotheliomas are a family of vascular neoplasms of intermediate malignancy.

Lentigo maligna is where melanocyte cells have become malignant and grow continuously along the stratum basale of the skin, but have not invaded below the epidermis. Lentigo maligna is not the same as lentigo maligna melanoma, as detailed below. It typically progresses very slowly and can remain in a non-invasive form for years.

Hidradenoma refers to a benign adnexal tumor of the apical sweat gland. These are 1–3 cm translucent blue cystic nodules. It usually presents as a single, small skin-colored lesion, and may be considered closely related to or a variant of poromas. Hidradenomas are often sub-classified based on subtle histologic differences, for example:

Extramammary Paget's disease (EMPD) is a rare and slow-growing malignancy which occurs within the epithelium and accounts for 6.5% of all Paget's disease. The clinical presentation of this disease is similar to the characteristics of mammary Paget's disease (MPD). However, unlike MPD, which occurs in large lactiferous ducts and then extends into the epidermis, EMPD originates in glandular regions rich in apocrine secretions outside the mammary glands. EMPD incidence is increasing by 3.2% every year, affecting hormonally-targeted tissues such as the vulva and scrotum. In women, 81.3% of EMPD cases are related to the vulva, while for men, 43.2% of the manifestations present at the scrotum.
Infantile digital fibromatosis (IDF), also termed inclusion body fibromatosis, Reye tumor, or Reye's tumor, usually occurs as a single, small, asymptomatic, nodule in the dermis on a finger or toe of infants and young children. IMF is a rare disorder with approximately 200 cases reported in the medical literature as of 2021. The World Health Organization in 2020 classified these nodules as a specific benign tumor type in the category of fibroblastic and myofibroblastic tumors. IDF was first described by the Australian pathologist, Douglas Reye, in 1965.
Atypical fibroxanthoma of the skin is a low-grade malignancy related to malignant fibrous histiocytoma, which it resembles histologically.
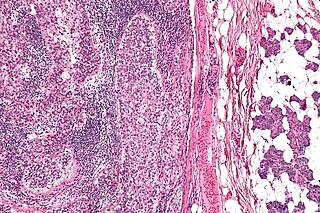
Sebaceous carcinoma, also known as sebaceous gland carcinoma (SGc), sebaceous cell carcinoma, and meibomian gland carcinoma is an uncommon malignant cutaneous tumor. Most are typically about 1.4 cm at presentation. SGc originates from sebaceous glands in the skin and, therefore, may originate anywhere in the body where these glands are found. SGc can be divided into 2 types: periocular and extraocular. The periocular region is rich in sebaceous glands making it a common site of origin. The cause of these lesions in the vast majority of cases is unknown. Occasional cases may be associated with Muir-Torre syndrome. SGc accounts for approximately 0.7% of all skin cancers, and the incidence of SGc is highest in Caucasian, Asian, and Indian populations. Due to the rarity of this tumor and variability in clinical and histological presentation, SGc is often misdiagnosed as an inflammatory condition or a more common neoplasm. SGc is commonly treated with wide local excision or Mohs micrographic surgery, and the relative survival rates at 5 and 10 years are 92.72 and 86.98%, respectively.

Poromas are rare, benign, cutaneous adnexal tumors. Cutaneous adnexal tumors are a group of skin tumors consisting of tissues that have differentiated towards one or more of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands. Poromas are eccrine or apocrine sweat gland tumors derived from the cells in the terminal portion of these glands' ducts. This part of the sweat gland duct is termed the acrosyringium and had led to grouping poromas in the acrospiroma class of skin tumors. Here, poromas are regarded as distinct sweat gland tumors that differ from other sweat gland tumors by their characteristic clinical presentations, microscopic histopathology, and the genetic mutations that their neoplastic cells have recently been found to carry.
Porocarcinoma (PCA) is a rare form of skin cancer that develops in eccrine sweat glands, i.e. the body's widely distributed major type of sweat glands, as opposed to the apocrine sweat glands which are located primarily in the armpits and perineal area. This cancer typically develops in individuals as a single cutaneous tumor in the intraepidermal spiral part of these sweat glands' ducts at or near to where they open on the skin's surface. PCA tumors are classified as one form of the cutaneous adnexal tumors; in a study of 2,205 cases, PCA was the most common (11.8%) form of these tumors.

Spiradenomas (SA) are rare, benign cutaneous adnexal tumors that may progress to become their malignant counterparts, i.e. spiradenocarcinomas (SAC). Cutaneous adnexal tumors are a group of skin tumors consisting of tissues that have differentiated towards one of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands. SA and SAC tumors were regarded as eccrine gland tumors and termed eccrine spiradenomas and eccrine spiradenocarcinomas, respectively. However, more recent studies have found them to be hair follicle tumors and commonly term them spiradenomas and spiradenocarcinomas, respectively. Further confusing the situation, SA-like and SAC-like tumors are also 1) manifestations of the inherited disorder, CYLD cutaneous syndrome (CCS), and 2) have repeatedly been confused with an entirely different tumor, adenoid cystic carcinomas of the salivary gland. Here, SA and SAC are strictly defined as sporadic hair follicle tumors that do not include the hereditary CCS spiradenomas and heridtary spiradenocarcinoms of CCS or the adenoid cystic carcinomas.
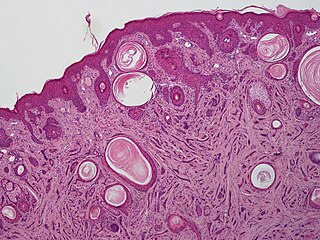
Microcystic adnexal carcinoma (MAC) is a rare sweat gland cancer, which often appears as a yellow spot or bump in the skin. It usually occurs in the neck or head, although cases have been documented in other areas of the body. Most diagnosis occur past the age of 50. Although considered an invasive cancer, metastasis rarely occurs. If the tumor spreads, it can grow and invade fat, muscles, and other types of tissue. Main treatments are wide local excision or Mohs micrographic surgery, which ensures that most, if not all, cancer cells are removed surgically.
Skin cancer, or neoplasia, is the most common type of cancer diagnosed in horses, accounting for 45 to 80% of all cancers diagnosed. Sarcoids are the most common type of skin neoplasm and are the most common type of cancer overall in horses. Squamous-cell carcinoma is the second-most prevalent skin cancer, followed by melanoma. Squamous-cell carcinoma and melanoma usually occur in horses greater than 9-years-old, while sarcoids commonly affect horses 3 to 6 years old. Surgical biopsy is the method of choice for diagnosis of most equine skin cancers, but is contraindicated for cases of sarcoids. Prognosis and treatment effectiveness varies based on type of cancer, degree of local tissue destruction, evidence of spread to other organs (metastasis) and location of the tumor. Not all cancers metastasize and some can be cured or mitigated by surgical removal of the cancerous tissue or through use of chemotherapeutic drugs.

Myoepithelioma of the head and neck, also myoepithelioma, is a salivary gland tumour of the head and neck that is usually benign. When malignant, which is exceedingly rare, they are known as malignant myoepithelioma or Myoepithelial carcinoma, and they account for 1% of the salivary tumors with poor prognosis.
Ectomesenchymal chondromyxoid tumor (ECT) is a benign intraoral tumor with presumed origin from undifferentiated (ecto)mesenchymal cells. There are some who think it is a myoepithelial tumor type.
Ovarian germ cell tumors (OGCTs) are heterogeneous tumors that are derived from the primitive germ cells of the embryonic gonad, which accounts for about 2.6% of all ovarian malignancies. There are four main types of OGCTs, namely dysgerminomas, yolk sac tumor, teratoma, and choriocarcinoma.

