Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.
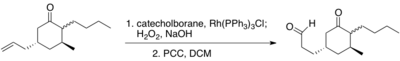
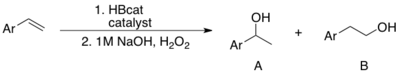
Hydroboration–oxidation reaction is a two-step hydration reaction that converts an alkene into an alcohol. The process results in the syn addition of a hydrogen and a hydroxyl group where the double bond had been. Hydroboration–oxidation is an anti-Markovnikov reaction, with the hydroxyl group attaching to the less-substituted carbon. The reaction thus provides a more stereospecific and complementary regiochemical alternative to other hydration reactions such as acid-catalyzed addition and the oxymercuration–reduction process. The reaction was first reported by Herbert C. Brown in the late 1950s and it was recognized in his receiving the Nobel Prize in Chemistry in 1979.
In organic chemistry, hydroformylation, also known as oxo synthesis or oxo process, is an industrial process for the production of aldehydes from alkenes. This chemical reaction entails the net addition of a formyl group and a hydrogen atom to a carbon-carbon double bond. This process has undergone continuous growth since its invention: production capacity reached 6.6×106 tons in 1995. It is important because aldehydes are easily converted into many secondary products. For example, the resultant aldehydes are hydrogenated to alcohols that are converted to detergents. Hydroformylation is also used in speciality chemicals, relevant to the organic synthesis of fragrances and pharmaceuticals. The development of hydroformylation is one of the premier achievements of 20th-century industrial chemistry.

Wilkinson's catalyst (chloridotris(triphenylphosphine)rhodium(I)) is a coordination complex of rhodium with the formula [RhCl(PPh3)], where 'Ph' denotes a phenyl group. It is a red-brown colored solid that is soluble in hydrocarbon solvents such as benzene, and more so in tetrahydrofuran or chlorinated solvents such as dichloromethane. The compound is widely used as a catalyst for hydrogenation of alkenes. It is named after chemist and Nobel laureate Sir Geoffrey Wilkinson, who first popularized its use.
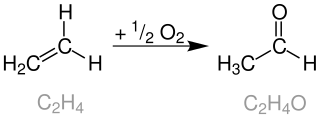
The Wacker process or the Hoechst-Wacker process refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride and copper(II) chloride as the catalyst. This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.

The Jacobsen epoxidation, sometimes also referred to as Jacobsen-Katsuki epoxidation is a chemical reaction which allows enantioselective epoxidation of unfunctionalized alkyl- and aryl- substituted alkenes. It is complementary to the Sharpless epoxidation (used to form epoxides from the double bond in allylic alcohols). The Jacobsen epoxidation gains its stereoselectivity from a C2 symmetric manganese(III) salen-like ligand, which is used in catalytic amounts. The manganese atom transfers an oxygen atom from chlorine bleach or similar oxidant. The reaction takes its name from its inventor, Eric Jacobsen, with Tsutomu Katsuki sometimes being included. Chiral-directing catalysts are useful to organic chemists trying to control the stereochemistry of biologically active compounds and develop enantiopure drugs.

Danishefsky's diene is an organosilicon compound and a diene with the formal name trans-1-methoxy-3-trimethylsilyloxy-buta-1,3-diene named after Samuel J. Danishefsky. Because the diene is very electron-rich it is a very reactive reagent in Diels-Alder reactions. This diene reacts rapidly with electrophilic alkenes, such as maleic anhydride. The methoxy group promotes highly regioselective additions. The diene is known to react with amines, aldehydes, alkenes and alkynes. Reactions with imines and nitro-olefins have been reported.
Hydrosilylation, also called catalytic hydrosilation, describes the addition of Si-H bonds across unsaturated bonds. Ordinarily the reaction is conducted catalytically and usually the substrates are unsaturated organic compounds. Alkenes and alkynes give alkyl and vinyl silanes; aldehydes and ketones give silyl ethers. Hydrosilylation has been called the "most important application of platinum in homogeneous catalysis."
Asymmetric catalytic oxidation is a technique of oxidizing various substrates to give an enantio-enriched product using a catalyst. Typically, but not necessarily, asymmetry is induced by the chirality of the catalyst. Typically, but again not necessarily, the methodology applies to organic substrates. Functional groups that can be prochiral and readily susceptible to oxidation include certain alkenes and thioethers. Challenging but pervasive prochiral substrates are C-H bonds of alkanes. Instead of introducing oxygen, some catalysts, biological and otherwise, enantioselectively introduce halogens, another form of oxidation.
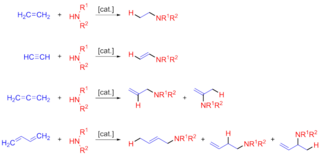
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.
Bis(oxazoline) ligands (often abbreviated BOX ligands) are a class of privileged chiral ligands containing two oxazoline rings. They are typically C2‑symmetric and exist in a wide variety of forms; with structures based around CH2 or pyridine linkers being particularly common (often generalised BOX and PyBOX respectively). The coordination complexes of bis(oxazoline) ligands are used in asymmetric catalysis. These ligands are examples of C2-symmetric ligands.
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being collectively awarded one half of the 2001 Nobel Prize in Chemistry.

Jacobsen's catalyst is the common name for N,N'-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminomanganese(III) chloride, a coordination compound of manganese and a salen-type ligand. It is used as an asymmetric catalyst in the Jacobsen epoxidation, which is renowned for its ability to enantioselectively transform prochiral alkenes into epoxides. Before its development, catalysts for the asymmetric epoxidation of alkenes required the substrate to have a directing functional group, such as an alcohol as seen in the Sharpless epoxidation. This compound has two enantiomers, which give the appropriate epoxide product from the alkene starting material.

DuPhos is a class of organophosphorus compound that are used ligands for asymmetric synthesis. The name DuPhos is derived from (1) the chemical company that sponsored the research leading to this ligand's invention, DuPont and (2) the compound is a diphosphine ligand type. Specifically it is classified as a C2-symmetric ligand, consisting of two phospholanes rings affixed to a benzene ring.
Enantioselective ketone reductions convert prochiral ketones into chiral, non-racemic alcohols and are used heavily for the synthesis of stereodefined alcohols.
Metal-catalyzed cyclopropanations are chemical reactions that result in the formation of a cyclopropane ring from a metal carbenoid species and an alkene. In the Simmons–Smith reaction the metal involved is zinc. Metal carbenoid species can be generated through the reaction of a diazo compound with a transition metal). The intramolecular variant of this reaction was first reported in 1961. Rhodium carboxylate complexes, such as dirhodium tetraacetate, are common catalysts. Enantioselective cyclopropanations have been developed.

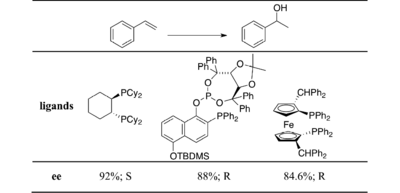
Phosphinooxazolines are a class of chiral ligands used in asymmetric catalysis. Colorless solids, PHOX ligands feature a tertiary phosphine group, often diphenyl, and an oxazoline ligand in the ortho position. The oxazoline, which carries the stereogenic center, coordinates through nitrogen, the result being that PHOX ligands are P,N-chelating ligands. Most phosphine ligands used in asymmetric catalysis are diphosphines, so the PHOX ligands are distinctive. Some evidence exists that PHOX ligands are hemilabile.
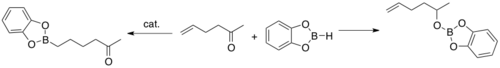
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.
Clark Landis is an American chemist, whose research focuses on organic and inorganic chemistry. He is currently a Professor of Chemistry at the University of Wisconsin–Madison. He was awarded the ACS Award in Organometallic Chemistry in 2010, and is a fellow of the American Chemical Society and the American Association for the Advancement of Science.
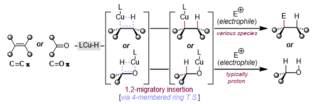
A hydrocupration is a chemical reaction whereby a ligated copper hydride species, reacts with a carbon-carbon or carbon-oxygen pi-system; this insertion is typically thought to occur via a four-membered ring transition state, producing a new copper-carbon or copper-oxygen sigma-bond and a stable (generally) carbon-hydrogen sigma-bond. In the latter instance (copper-oxygen), protonation (protodemetalation) is typical – the former (copper-carbon) has broad utility. The generated copper-carbon bond (organocuprate) has been employed in various nucleophilic additions to polar conjugated and non-conjugated systems and has also been used to forge new carbon-heteroatom bonds.