Molecular autopsy or postmortem molecular testing is a set of molecular techniques used in forensic medicine to attempt to determine the cause of death in unexplained cases, in particular sudden unexplained deaths (for example sudden cardiac death). About 30% of sudden cardiac deaths in young people are not explained after full conventional autopsy, and are classified as sudden unexplained deaths. The use of a panel of genetic markers for long QT syndrome, catecholaminergic polymorphic ventricular tachycardia and cardiac channel myopathies elucidated around 40 to 45% of the cases.[1]
In today’s day and age the use of Molecular Autopsy has come with its share of ethical issues. The issues are raised because there are no set laws that a Medical Examiner must follow. For instance it is not required for an examiner to get permission from a relative to go forth with a molecular autopsy. This has created many issues for families because they may not always want to know the results of why a loved one died. Knowing this information can create anxiety and concern for family members over a possible mutation of their own gene that could cause their death, while they would have no way of stopping it. It also creates an issue because with most if not all examinations, samples of a test are retained in a lab. This means the tests from a loved one is saved forever, to be possibly used in a different experiment. The family usually has no say on whether this will happen or not.
The problem that arises for medical examiners is that if an examination is done and the lives of family members could be at risk, they have no authorization to tell the family if they do not wish to know. Some examiners believe that this is against their duty as a professional doctor. For example, it has been estimated that 30% of young sudden cardiac deaths can be traced to being inherited. So doctors feel that it is against their profession to not let someone know when they could be at risk.[2]
Methods
When a traditional medical autopsy is not able to determine the sudden cause of death, molecular autopsy may help provide an alternative insight through the use of Deoxyribonucleic acid (DNA) sequencing. It looks at things from a cellular level instead of only what the human eye can see.
The first step in performing a molecular autopsy is to obtain a sample of blood or tissue from the individual after death has occurred. DNA is then extracted from the blood sample in order to undergo a process of genetic sequencing. Then, the DNA sequence is carefully analyzed to detect any gene mutations that may be a cause of sudden death. Initially, molecular autopsy focused on the direct DNA sequencing of four genes. However, recent advancements in sequencing technologies have made it possible to screen a large number of genes at once from a small sample of DNA through whole-exome sequencing (WES) in which the coding regions of all 22,000 genes are sequenced. This potentially allows the detection of genetic variants of genes related to all major diseases.[3]
Case Studies
A study of sudden death brought a mother to once question whether her thirteen-year-old son has what previously killed her seventeen-year-old son. This son had been found lying in bed dead with an autopsy that was inconclusive. Many blamed it on drug use and abuse, but that was really not the cause.
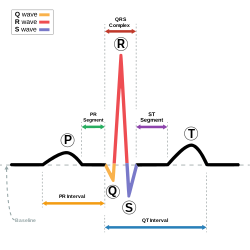
PQRST complex of a heart beat
Almost half the sudden deaths of previously healthy children have no findings on autopsy. These children are referred to as having sudden unexplained death syndrome (SUDS). In the Olmsted County population study, six of the twelve cases died of unknown causes of SUDS. A lot of forensic pathologists blame a fatal arrhythmia of the heart to be the cause of SUDS due to the lethal disorders like long QT syndrome (LQTS). This is a prolonged QT interval in the heart’s natural rhythm. This is can leave no trace for an autopsy. The clinical signs of LQTS are syncope, seizures, or sudden death.
In England there are around 200 SUDS cases yearly, and nearly a third of those were blamed on LQTS. This however, cannot be proved without an electrocardiogram before death.
By looking at the molecular level of the issues that cause SUDS and/or LQTS, they may be able to find the ion channels that are cardiac defective. There are six LQTS genetic markers, five LQTS genes, and around 200 mutations identified all in patients with LQTS. By targeting these molecules, molecular autopsy can be possible. This is how molecular autopsy is relevant in all three of the following cases.
Case 1
In this case of the mother with questions of her living son possible having the same issue that her now dead son had, there was a history of these LQTS clinical signs that were stated above in the family. Specifically, the grandmother had syncopal episodes multiple times. Although, multiple electrocardiograms showed no significant findings that would lead to a diagnosis of LQTS.
There were multiple studies done, one in particular was the epinephrine-triggered alterations in repolarization. This showed the results of having five nucleotides (guanine [g], cytosine [c], guanine, cytosine, and thymidine [t]) from positions 735 - 739 were not present. These are the genetic components of DNA. This resulted in the cardiac potassium channel to cause a shift of amino acids. This shift is where the stop codon at an amino acid is introduced and needed. This can severely impact the depolarization and repolarization of the heart, which is crucial for the normal rhythm of the muscle.[4]
Case 2
Another study was done for molecular autopsy on the RyR2-encoded cardiac ryanodine receptor in SUDS. There were 49 cases in this study, 30 of which were male. Thirteen of the 49 studied had a family history of syncope. In seven of these cases of SUDS, there were six distinct RyR2 missense mutations. During these deaths, the activities were as follows: three cases of exertion, one case of emotion, and three unknown cases. This study was of the first on RyR2 in molecular autopsy. It targeted 18 of the 105 protein-encoding exons of the cardiac ryanodine receptor/calcium release channel. This revealed one in every seven to be positive for the RyR2 mutations in SUDS. This studied showed that testing of this mutation should be a part of the autopsy investigation. This study also proved that this mutation is possibly inheritable.[5]
Case 3
Another study is the pharmacogenomics as molecular autopsy for forensic toxicology. This study is looking at the genotyping of cytochrome P450 3A4*1B and 3A5*3. Pharmacogenetics is the study of genetic contributions to drug action. This can help in certifying a fentanyl toxicity. Fentanyl is used for anesthesia in surgery or pain control/management in animals and humans. This drug can have variable metabolisms due to the different alleles in the cytochrome P450. This study looked at 25 different fentanyl related deaths (22 caucasians, 1 African American, and 2 Native Americans). from the Milwaukee county medical Examiner’s office and referrals. Blood was taken and analyzed after death by radioimmunoassay and liquid chromatography/mass spectrometry. This study showed the average fentanyl concentration in CYP3A4*1B wild type and 3A5*3 homozygous variant cases were higher than those of the CYP3A4*1B variant cases (this was not a significant difference). The data taken from this study gave scientific evidence that CYP3A5 is involved in the fentanyl metabolism, where as the homozygous CYP3A5*3 causes impaired metabolism of fentanyl. Genotyping CYP3A4*1B and 3A5*3 variants may help to certify the fentanyl toxicity. For further studying of this subject, there will be more cases needed. This study was mainly to supply information for this drug monitoring and pain management.[6]
Relationship with molecular autopsy
Molecular autopsy has become a huge component in the investigation process of SUD, specifically sudden cardiac death (SCD). The causes of SCD range widely but the greatest contributor to SCD is an underlying genetic predisposition, especially in those under the age of 40. The inherited diseases include, but are not limited to, primary arrythmogenic disorders and inherited cardiomyopathies. Molecular autopsy not only helps identify an explanation for SUD, but evaluates the potential risks that relatives may have in relation to cardiovascular disease. Over 3 million people die of SCD a year, making molecular autopsy for SCD in high demand. Using molecular autopsy for SCD in the young, fit, and seemingly healthy individual is an increasingly interesting topic for research. Up to 30% of the autopsies given post-mortem to young individuals who die of SCD have no cause of death identified, called autopsy-negative or sudden arrhythmic death syndrome (SADS). This is because many primary arrhythmogenic disorders do not cause structural damage to the heart, making it difficult for pathologists to draw a conclusion on the cause of death.
Genetic testing for SADS cases started over ten years ago. A sample of the cadaver’s blood is taken and tested. The molecular autopsy focuses on four main genes: KCNQ1, KCNH2, SCN5A, and RYR2. Greater than 95% of the mutations found in the molecular autopsy are a chromosome dominant trait, indicating that half of the children to the tested individual also carry the mutated gene.[7]
↑ McGuire, Amy L., Quianta Moore, Mary Majumder, Magdalena Walkiewicz, Christine M. Eng, John W. Belmont, Salma Nassef, Sandra Darilek, Katie Rutherford, Stacey Pereira, Steven E. Scherer, V. Reid Sutton, Dwayne Wolf, Richard A. Gibbs, Roger Kahn, Luis A. Sanchez, and The Molecular Autopsy Consortium of Houston (MATCH). "The Ethics of Conducting Molecular Autopsies in Cases of Sudden Death in the Young." The Ethics of Conducting Molecular Autopsies in Cases of Sudden Death in the Young (2016): n. pag. Genome Research. Cold Spring Harbor Laboratory Press, Sept. 2016. Web. 21 Apr. 2017.
↑ Lahrouchi, Najim, Elijah R. Behr, and Connie R. Bezzina. “Next-Generation Sequencing in Post-Mortem Genetic Testing of Young Sudden Cardiac Death Cases.” Frontiers in Cardiovascular Medicine 3 (2016): 13. PMC. Web. 2 Apr. 2017.
↑ Ackerman, Michael J., MD, PhD, David J. Tester, BS, and David J. Driscoll, MD. "Molecular Autopsy of Sudden Unexplained Death in the Young: The American Journal of Forensic Medicine and Pathology." LWW. American Journal of Forensic Medicine and Pathology, June 2001. Web. 20 Mar. 2017.
↑ Tester, David J., BS, Daniel B. Spoon, BS, Hector H. Valdivia, MD, PhD, Jonathan C. Makelski, MD, and Michael J. Ackerman, MD, PhD. "Targeted Mutational Analysis of the RyR2-Encoded Cardiac Ryanodine Receptor in Sudden Unexplained Death: A Molecular Autopsy of 49 Medical Examiner/Coroner's Cases." May Clinic Proceedings. N.p., 2004. Web.
↑ Jin, Ming, Susan B. Gock, Paul J. Jannetto, Jeffrey M. Jentzen, and Steven H. Wong. "Pharmacogenomic as Molecular Autopsy for Forensic Toxicology: Genotyping Cytochrome P450 3A4*1B and 3A5*3 for 25 Fentanyl Cases." Journal of Analytical Toxicology. N.p., 2005. Web.
↑ Semsarian, Christopher, and Robert M. Hamilton. (2012) "Key Role of the Molecular Autopsy in Sudden Unexpected Death." Heart Rhythm 9.1: 145-50. doi: 10.1016/j.hrthm.2011.07.034. Semsarian, C., Ingles, J. and Wilde, A.A. (2015) Sudden Cardiac Death in the Young: The Molecular Autopsy and a Practical Approach to Surviving Relatives. European Heart Journal, 36 (21), 1290-1296. Doi: 10.1093/eurheartj/ehv063
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.