
Eosinophilia is a condition in which the eosinophil count in the peripheral blood exceeds 5×108/L (500/μL). Hypereosinophilia is an elevation in an individual's circulating blood eosinophil count above 1.5 × 109/L (i.e. 1,500/μL). The hypereosinophilic syndrome is a sustained elevation in this count above 1.5 × 109/L (i.e. 1,500/μL) that is also associated with evidence of eosinophil-based tissue injury.

Fibrosis, also known as fibrotic scarring, is a pathological wound healing in which connective tissue replaces normal parenchymal tissue to the extent that it goes unchecked, leading to considerable tissue remodelling and the formation of permanent scar tissue.

Rituximab, sold under the brand name Rituxan among others, is a monoclonal antibody medication used to treat certain autoimmune diseases and types of cancer. It is used for non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, idiopathic thrombocytopenic purpura, pemphigus vulgaris, myasthenia gravis and Epstein–Barr virus-positive mucocutaneous ulcers. It is given by slow intravenous infusion. Biosimilars of Rituxan include Blitzima, Riabni, Ritemvia, Rituenza, Rixathon, Ruxience, and Truxima.
Neuromyelitis optica spectrum disorders (NMOSD), including neuromyelitis optica (NMO), are autoimmune diseases characterized by acute inflammation of the optic nerve and the spinal cord (myelitis). Episodes of ON and myelitis can be simultaneous or successive. A relapsing disease course is common, especially in untreated patients. In more than 80% of cases, NMO is caused by immunoglobulin G autoantibodies to aquaporin 4 (anti-AQP4), the most abundant water channel protein in the central nervous system. A subset of anti-AQP4-negative cases is associated with antibodies against myelin oligodendrocyte glycoprotein (anti-MOG). Rarely, NMO may occur in the context of other autoimmune diseases or infectious diseases. In some cases, the etiology remains unknown.

Hypersensitivity pneumonitis (HP) or extrinsic allergic alveolitis (EAA) is a syndrome caused by the repetitive inhalation of antigens from the environment in susceptible or sensitized people. Common antigens include molds, bacteria, bird droppings, bird feathers, agricultural dusts, bioaerosols and chemicals from paints or plastics. People affected by this type of lung inflammation (pneumonitis) are commonly exposed to the antigens by their occupations, hobbies, the environment and animals. The inhaled antigens produce a hypersensitivity immune reaction causing inflammation of the airspaces (alveoli) and small airways (bronchioles) within the lung. Hypersensitivity pneumonitis may eventually lead to interstitial lung disease.

Livedo reticularis is a common skin finding consisting of a mottled reticulated vascular pattern that appears as a lace-like purplish discoloration of the skin. The discoloration is caused by reduction in blood flow through the arterioles that supply the cutaneous capillaries, resulting in deoxygenated blood showing as blue discoloration. This can be a secondary effect of a condition that increases a person's risk of forming blood clots, including a wide array of pathological and nonpathological conditions. Examples include hyperlipidemia, microvascular hematological or anemia states, nutritional deficiencies, hyper- and autoimmune diseases, and drugs/toxins.
Retroperitoneal fibrosis or Ormond's disease is a disease featuring the proliferation of fibrous tissue in the retroperitoneum, the compartment of the body containing the kidneys, aorta, renal tract, and various other structures. It may present with lower back pain, kidney failure, hypertension, deep vein thrombosis, and other obstructive symptoms. It is named after John Kelso Ormond, who rediscovered the condition in 1948.
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an acquired autoimmune disease of the peripheral nervous system characterized by progressive weakness and impaired sensory function in the legs and arms. The disorder is sometimes called chronic relapsing polyneuropathy (CRP) or chronic inflammatory demyelinating polyradiculoneuropathy. CIDP is closely related to Guillain–Barré syndrome and it is considered the chronic counterpart of that acute disease. Its symptoms are also similar to progressive inflammatory neuropathy. It is one of several types of neuropathy.
Autoimmune Pancreatitis (AIP) is an increasingly recognized type of chronic pancreatitis that can be difficult to distinguish from pancreatic carcinoma but which responds to treatment with corticosteroids, particularly prednisone. Although autoimmune pancreatitis is quite rare, it constitutes an important clinical problem for both patients and their clinicians: the disease commonly presents itself as a tumorous mass which is diagnostically indistinguishable from pancreatic cancer, a disease that is much more common in addition to being very dangerous. Hence, some patients undergo pancreatic surgery, which is associated to substantial mortality and morbidity, out of the fear by patients and clinicians to undertreat a malignancy. However, surgery is not a good treatment for this condition as AIP responds well to immunosuppressive treatment. There are two categories of AIP: Type 1 and Type 2, each with distinct clinical profiles.
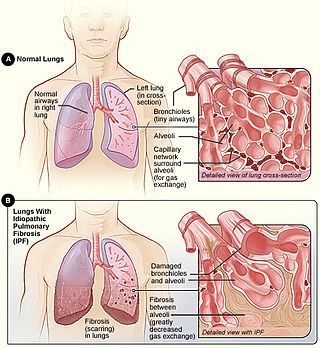
Idiopathic pulmonary fibrosis (IPF), or (formerly) fibrosing alveolitis, is a rare, progressive illness of the respiratory system, characterized by the thickening and stiffening of lung tissue, associated with the formation of scar tissue. It is a type of chronic scarring lung disease characterized by a progressive and irreversible decline in lung function. The tissue in the lungs becomes thick and stiff, which affects the tissue that surrounds the air sacs in the lungs. Symptoms typically include gradual onset of shortness of breath and a dry cough. Other changes may include feeling tired, and abnormally large and dome shaped finger and toenails. Complications may include pulmonary hypertension, heart failure, pneumonia or pulmonary embolism.
Riedel's thyroiditis, is a chronic form of thyroiditis. It is now believed that Riedel's thyroiditis is one manifestation of a systemic disease that can affect many organ systems called IgG4-related disease. It is often a multi-organ disease affecting pancreas, liver, kidney, salivary and orbital tissues and retroperitoneum. The hallmarks of the disease are fibrosis and infiltration by IgG4 secreting plasma cells.

Angiolymphoid hyperplasia with eosinophilia usually presents with pink to red-brown, dome-shaped, dermal papules or nodules of the head or neck, especially about the ears and on the scalp.
Granulomatous mastitis can be divided into idiopathic granulomatous mastitis and granulomatous mastitis occurring as a rare secondary complication of a great variety of other conditions such as tuberculosis and other infections, sarcoidosis and granulomatosis with polyangiitis. Special forms of granulomatous mastitis occur as complication of diabetes. Some cases are due to silicone injection or other foreign body reactions.

Idiopathic sclerosing mesenteritis (ISM) is a rare disease of the small intestine, characterized by chronic inflammation and eventual fibrosis of the mesentery. It has also been called mesenteric lipodystrophy, or retractile mesenteritis.

IgG4-related disease (IgG4-RD), formerly known as IgG4-related systemic disease, is a chronic inflammatory condition characterized by tissue infiltration with lymphocytes and IgG4-secreting plasma cells, various degrees of fibrosis (scarring) and a usually prompt response to oral steroids. In approximately 51–70% of people with this disease, serum IgG4 concentrations are elevated during an acute phase.
Chronic sclerosing sialadenitis is a chronic (long-lasting) inflammatory condition affecting the salivary gland. Relatively rare in occurrence, this condition is benign, but presents as hard, indurated and enlarged masses that are clinically indistinguishable from salivary gland neoplasms or tumors. It is now regarded as a manifestation of IgG4-related disease.
IgG4-related prostatitis is prostate involvement in men with IgG4-related disease (IgG4-RD), which is an emerging fibroinflammatory disease entity which is characterised (i) by a tendency to mass forming lesions in multiple sites of the body and (ii) by usually a prompt response to steroid therapy.

IgG4-related ophthalmic disease (IgG4-ROD) is the recommended term to describe orbital manifestations of the systemic condition IgG4-related disease, which is characterised by infiltration of lymphocytes and plasma cells and subsequent fibrosis in involved structures. It can involve one or more of the orbital structures.
IgG4-related skin disease is the recommended name for skin manifestations in IgG4-related disease (IgG4-RD). Multiple different skin manifestations have been described.
