
Prune belly syndrome is a rare, genetic birth defect affecting about 1 in 40,000 births. About 97% of those affected are male. Prune belly syndrome is a congenital disorder of the urinary system, characterized by a triad of symptoms. The syndrome is named for the mass of wrinkled skin that is often present on the abdomen of those with the disorder.

Tetralogy of Fallot (TOF), formerly known as Steno-Fallot tetralogy, is a congenital heart defect characterized by four specific cardiac defects. Classically, the four defects are:

Patent ductus arteriosus (PDA) is a medical condition in which the ductus arteriosus fails to close after birth: this allows a portion of oxygenated blood from the left heart to flow back to the lungs through the aorta, which has a higher blood pressure, to the pulmonary artery, which has a lower blood pressure. Symptoms are uncommon at birth and shortly thereafter, but later in the first year of life there is often the onset of an increased work of breathing and failure to gain weight at a normal rate. With time, an uncorrected PDA usually leads to pulmonary hypertension followed by right-sided heart failure.

The ductus arteriosus, also called the ductus Botalli, named after the Italian physiologist Leonardo Botallo, is a blood vessel in the developing fetus connecting the trunk of the pulmonary artery to the proximal descending aorta. It allows most of the blood from the right ventricle to bypass the fetus's fluid-filled non-functioning lungs. Upon closure at birth, it becomes the ligamentum arteriosum.

A congenital heart defect (CHD), also known as a congenital heart anomaly, congenital cardiovascular malformation, and congenital heart disease, is a defect in the structure of the heart or great vessels that is present at birth. A congenital heart defect is classed as a cardiovascular disease. Signs and symptoms depend on the specific type of defect. Symptoms can vary from none to life-threatening. When present, symptoms are variable and may include rapid breathing, bluish skin (cyanosis), poor weight gain, and feeling tired. CHD does not cause chest pain. Most congenital heart defects are not associated with other diseases. A complication of CHD is heart failure.
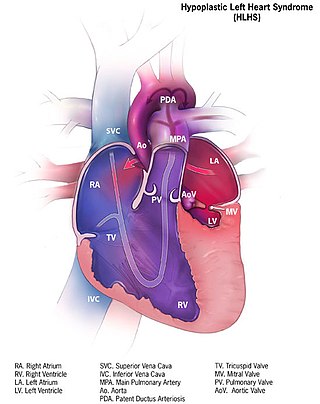
Hypoplastic left heart syndrome (HLHS) is a rare congenital heart defect in which the left side of the heart is severely underdeveloped and incapable of supporting the systemic circulation. It is estimated to account for 2-3% of all congenital heart disease. Early signs and symptoms include poor feeding, cyanosis, and diminished pulse in the extremities. The etiology is believed to be multifactorial resulting from a combination of genetic mutations and defects resulting in altered blood flow in the heart. Several structures can be affected including the left ventricle, aorta, aortic valve, or mitral valve all resulting in decreased systemic blood flow.

Persistent truncus arteriosus (PTA), often referred to simply as truncus arteriosus, is a rare form of congenital heart disease that presents at birth. In this condition, the embryological structure known as the truncus arteriosus fails to properly divide into the pulmonary trunk and aorta. This results in one arterial trunk arising from the heart and providing mixed blood to the coronary arteries, pulmonary arteries, and systemic circulation. For the International Classification of Diseases (ICD-11), the International Paediatric and Congenital Cardiac Code (IPCCC) was developed to standardize the nomenclature of congenital heart disease. Under this system, English is now the official language, and persistent truncus arteriosus should properly be termed common arterial trunk.

Larsen syndrome (LS) is a congenital disorder discovered in 1950 by Larsen and associates when they observed dislocation of the large joints and face anomalies in six of their patients. Patients with Larsen syndrome normally present with a variety of symptoms, including congenital anterior dislocation of the knees, dislocation of the hips and elbows, flattened facial appearance, prominent foreheads, and depressed nasal bridges. Larsen syndrome can also cause a variety of cardiovascular and orthopedic abnormalities. This rare disorder is caused by a genetic defect in the gene encoding filamin B, a cytoplasmic protein that is important in regulating the structure and activity of the cytoskeleton. The gene that influences the emergence of Larsen syndrome is found in chromosome region, 3p21.1-14.1, a region containing human type VII collagen gene. Larsen syndrome has recently been described as a mesenchyme disorder that affects the connective tissue of an individual. Autosomal dominant and recessive forms of the disorder have been reported, although most cases are autosomal dominant. Reports have found that in Western societies, Larsen syndrome can be found in one in every 100,000 births, but this is most likely an underestimate because the disorder is frequently unrecognized or misdiagnosed.
Interrupted aortic arch is a very rare heart defect in which the aorta is not completely developed. There is a gap between the ascending and descending thoracic aorta. In a sense it is the complete form of a coarctation of the aorta. Almost all patients also have other cardiac anomalies, including a ventricular septal defect (VSD), aorto-pulmonary window, and truncus arteriosus. There are three types of interrupted aortic arch, with type B being the most common. Interrupted aortic arch is often associated with DiGeorge syndrome.
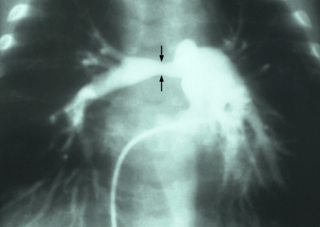
Pulmonary artery stenosis (PAS) is a narrowing of the pulmonary artery. The pulmonary artery is a blood vessel moving blood from the right side of the heart to the lungs. This narrowing can be due to many causes, including infection during pregnancy, a congenital heart defect, a problem with blood clotting in childhood or early adulthood, or a genetic change.

Familial aortic dissection or FAD refers to the splitting of the wall of the aorta in either the arch, ascending or descending portions. FAD is thought to be passed down as an autosomal dominant disease and once inherited will result in dissection of the aorta, and dissecting aneurysm of the aorta, or rarely aortic or arterial dilation at a young age. Dissection refers to the actual tearing open of the aorta. However, the exact gene(s) involved has not yet been identified. It can occur in the absence of clinical features of Marfan syndrome and of systemic hypertension. Over time this weakness, along with systolic pressure, results in a tear in the aortic intima layer thus allowing blood to enter between the layers of tissue and cause further tearing. Eventually complete rupture of the aorta occurs and the pleural cavity fills with blood. Warning signs include chest pain, ischemia, and hemorrhaging in the chest cavity. This condition, unless found and treated early, usually results in death. Immediate surgery is the best treatment in most cases. FAD is not to be confused with PAU and IMH, both of which present in ways similar to that of familial aortic dissection.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.

Cantú syndrome is a rare condition characterized by hypertrichosis, osteochondrodysplasia, and cardiomegaly. Fewer than 50 cases have been described in the literature; they are associated with a mutation in the ABCC9-gene that codes for the ABCC9-protein.
Myhre syndrome (MS) is an ultrarare genetic disorder caused by dominant gain-of-function (GOF) mutations in the SMAD4 gene. MS mutations are missense heterozygous mutations affecting only Ile500 or Arg496 residues of the SMAD4 protein. MS patients exhibit manifestations of connective tissue disease including dysfunction of the integumentary, cardiovascular, respiratory, gastrointestinal, and musculoskeletal systems and is often characterized by proliferative systemic fibrosis. Some of these features are life threatening, such as airway or arterial narrowing and fibroproliferation of tissues including lung, heart, and liver. Consistent with these clinical observations, cells isolated from patients with MS demonstrate increased TGF-β signaling.

Absent pulmonary valve syndrome(APVS) is a congenital heart defect that occurs when the flaps of the pulmonary valve do not develop or are severely underdeveloped (hypoplasia) resulting in aneurysms (dilation) of the pulmonary arteries and softening of the trachea and bronchi (tracheobronchomalacia). Usually, APVS occurs together with other congenital heart defects, most commonly ventricular septal defect and right ventricular outflow tract obstruction. It is sometimes considered a variant of Tetralogy of Fallot. The first case of absent pulmonary valve syndrome was reported Crampton in 1830.
Char syndrome is an autosomal dominant congenital disease caused by mutations in TFAP2B gene which affects the development of the bones of the face as well as the heart and limbs. During embryo development, TFAP2B regulates the production of the protein AP-2β, a transcription factor that is active in the neural crest and helps regulate genes that control cell division and apoptosis. There are at least 10 mutations of this gene that have been identified in people presenting Char syndrome, which alters specific regions of the gene preventing production of the transcription factor and disrupting normal development of embryo structures. People with this condition present a very distinct facial appearance with flattened cheek bones, flat and broad tip nose, shortened distance between the nose and upper lip, triangular-shaped mouth with tick lips and strabismus. It is also characterized by a patent ductus arteriosus, which is the failure to close the ductus that connects the aorta and pulmonary artery during pre-birth life and may cause many symptoms including breathing issues and heart failure. Abnormalities of hand and finger development have also been reported in people with this condition, including short or absent fifth finger. Other abnormal findings include supernumerary nipples. These conditions often affect multiple members of a family and there are no reports of non-genetic factors that might be related with incidence of this syndrome. It was first described by Florence Char in 1978.
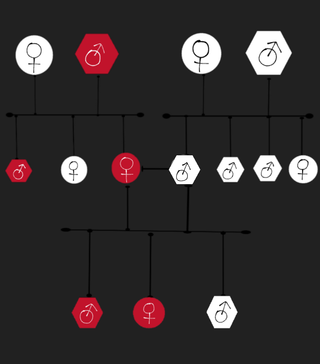
Familial thoracic aortic aneurysm and aortic dissection is a very rare vascular genetic disorder, it's characterized by recurrent thoracic aortic aneurysms and aortic dissections within a family, these mentioned complications affect one or more aortic segments without any other disease being associated with them. People with this disorder have a higher chance of having a potentially fatal aortic rupture. This disorder is the cause of 20% of thoracic aortic aneurysms
Meacham syndrome is a rare genetic disorder which is characterized by lung, diaphragmatic and genitourinary anomalies.

Pulmonary atresia with ventricular septal defect is a rare birth defect characterized by pulmonary valve atresia occurring alongside a defect on the right ventricular outflow tract.