An allele is a variation of the same sequence of nucleotides at the same place on a long DNA molecule, as described in leading textbooks on genetics and evolution.
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.

In genetics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome. Although certain definitions require the substitution to be present in a sufficiently large fraction of the population, many publications do not apply such a frequency threshold.
A haplotype is a group of alleles in an organism that are inherited together from a single parent.
In electrical engineering, statistical computing and bioinformatics, the Baum–Welch algorithm is a special case of the expectation–maximization algorithm used to find the unknown parameters of a hidden Markov model (HMM). It makes use of the forward-backward algorithm to compute the statistics for the expectation step.
In knot theory, there are several competing notions of the quantity writhe, or . In one sense, it is purely a property of an oriented link diagram and assumes integer values. In another sense, it is a quantity that describes the amount of "coiling" of a mathematical knot in three-dimensional space and assumes real numbers as values. In both cases, writhe is a geometric quantity, meaning that while deforming a curve in such a way that does not change its topology, one may still change its writhe.
A genetic marker is a gene or DNA sequence with a known location on a chromosome that can be used to identify individuals or species. It can be described as a variation that can be observed. A genetic marker may be a short DNA sequence, such as a sequence surrounding a single base-pair change, or a long one, like minisatellites.

In biology, a substitution model, also called models of DNA sequence evolution, are Markov models that describe changes over evolutionary time. These models describe evolutionary changes in macromolecules represented as sequence of symbols. Substitution models are used to calculate the likelihood of phylogenetic trees using multiple sequence alignment data. Thus, substitution models are central to maximum likelihood estimation of phylogeny as well as Bayesian inference in phylogeny. Estimates of evolutionary distances are typically calculated using substitution models. Substitution models are also central to phylogenetic invariants since they can be used to predict the frequencies of site pattern frequencies given a tree topology. Substitution models are necessary to simulate sequence data for a group of organisms related by a specific tree.
In mathematics, subshifts of finite type are used to model dynamical systems, and in particular are the objects of study in symbolic dynamics and ergodic theory. They also describe the set of all possible sequences executed by a finite state machine. The most widely studied shift spaces are the subshifts of finite type.

AFLP-PCR or just AFLP is a PCR-based tool used in genetics research, DNA fingerprinting, and in the practice of genetic engineering. Developed in the early 1990s by KeyGene, AFLP uses restriction enzymes to digest genomic DNA, followed by ligation of adaptors to the sticky ends of the restriction fragments. A subset of the restriction fragments is then selected to be amplified. This selection is achieved by using primers complementary to the adaptor sequence, the restriction site sequence and a few nucleotides inside the restriction site fragments. The amplified fragments are separated and visualized on denaturing on agarose gel electrophoresis, either through autoradiography or fluorescence methodologies, or via automated capillary sequencing instruments.

Genetic distance is a measure of the genetic divergence between species or between populations within a species, whether the distance measures time from common ancestor or degree of differentiation. Populations with many similar alleles have small genetic distances. This indicates that they are closely related and have a recent common ancestor.
The fixation index (FST) is a measure of population differentiation due to genetic structure. It is frequently estimated from genetic polymorphism data, such as single-nucleotide polymorphisms (SNP) or microsatellites. Developed as a special case of Wright's F-statistics, it is one of the most commonly used statistics in population genetics.

Masatoshi Nei is a Japanese-born American evolutionary biologist currently affiliated with the Department of Biology at Temple University as a Carnell Professor. He was, until recently, Evan Pugh Professor of Biology at Pennsylvania State University and Director of the Institute of Molecular Evolutionary Genetics; he was there from 1990 to 2015.
Bayesian inference of phylogeny combines the information in the prior and in the data likelihood to create the so-called posterior probability of trees, which is the probability that the tree is correct given the data, the prior and the likelihood model. Bayesian inference was introduced into molecular phylogenetics in the 1990s by three independent groups: Bruce Rannala and Ziheng Yang in Berkeley, Bob Mau in Madison, and Shuying Li in University of Iowa, the last two being PhD students at the time. The approach has become very popular since the release of the MrBayes software in 2001, and is now one of the most popular methods in molecular phylogenetics.
A number of different Markov models of DNA sequence evolution have been proposed. These substitution models differ in terms of the parameters used to describe the rates at which one nucleotide replaces another during evolution. These models are frequently used in molecular phylogenetic analyses. In particular, they are used during the calculation of likelihood of a tree and they are used to estimate the evolutionary distance between sequences from the observed differences between the sequences.
In population genetics, fixation is the change in a gene pool from a situation where there exists at least two variants of a particular gene (allele) in a given population to a situation where only one of the alleles remains. In the absence of mutation or heterozygote advantage, any allele must eventually be lost completely from the population or fixed. Whether a gene will ultimately be lost or fixed is dependent on selection coefficients and chance fluctuations in allelic proportions. Fixation can refer to a gene in general or particular nucleotide position in the DNA chain (locus).
Tajima's D is a population genetic test statistic created by and named after the Japanese researcher Fumio Tajima. Tajima's D is computed as the difference between two measures of genetic diversity: the mean number of pairwise differences and the number of segregating sites, each scaled so that they are expected to be the same in a neutrally evolving population of constant size.
Diversity Arrays Technology (DArT) is a high-throughput genetic marker technique that can detect allelic variations to provides comprehensive genome coverage without any DNA sequence information for genotyping and other genetic analysis. The general steps involve reducing the complexity of the genomic DNA with specific restriction enzymes, choosing diverse fragments to serve as representations for the parent genomes, amplify via polymerase chain reaction (PCR), insert fragments into a vector to be placed as probes within a microarray, then fluorescent targets from a reference sequence will be allowed to hybridize with probes and put through an imaging system. The objective is to identify and quantify various forms of DNA polymorphism within genomic DNA of sampled species.
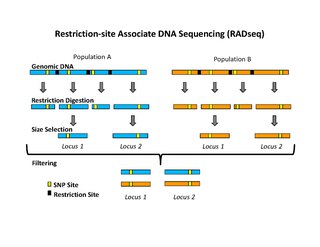
Restriction site associated DNA (RAD) markers are a type of genetic marker which are useful for association mapping, QTL-mapping, population genetics, ecological genetics and evolutionary genetics. The use of RAD markers for genetic mapping is often called RAD mapping. An important aspect of RAD markers and mapping is the process of isolating RAD tags, which are the DNA sequences that immediately flank each instance of a particular restriction site of a restriction enzyme throughout the genome. Once RAD tags have been isolated, they can be used to identify and genotype DNA sequence polymorphisms mainly in form of single nucleotide polymorphisms (SNPs). Polymorphisms that are identified and genotyped by isolating and analyzing RAD tags are referred to as RAD markers. Although genotyping by sequencing presents an approach similar to the RAD-seq method, they differ in some substantial ways.
Amino acid replacement is a change from one amino acid to a different amino acid in a protein due to point mutation in the corresponding DNA sequence. It is caused by nonsynonymous missense mutation which changes the codon sequence to code other amino acid instead of the original.







