Related Research Articles

Software testing is the act of checking whether software satisfies expectations.
The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules such as proteins and nucleic acids, which is overseen by the Worldwide Protein Data Bank (wwPDB). These structural data are obtained and deposited by biologists and biochemists worldwide through the use of experimental methodologies such as X-ray crystallography, NMR spectroscopy, and, increasingly, cryo-electron microscopy. All submitted data are reviewed by expert biocurators and, once approved, are made freely available on the Internet under the CC0 Public Domain Dedication. Global access to the data is provided by the websites of the wwPDB member organisations.

In statistics, an outlier is a data point that differs significantly from other observations. An outlier may be due to a variability in the measurement, an indication of novel data, or it may be the result of experimental error; the latter are sometimes excluded from the data set. An outlier can be an indication of exciting possibility, but can also cause serious problems in statistical analyses.
An HTML editor is a program used for editing HTML, the markup of a web page. Although the HTML markup in a web page can be controlled with any text editor, specialized HTML editors can offer convenience, added functionality, and organisation. For example, many HTML editors handle not only HTML, but also related technologies such as CSS, XML and JavaScript or ECMAScript. In some cases they also manage communication with remote web servers via FTP and WebDAV, and version control systems such as Subversion or Git. Many word processing, graphic design and page layout programs that are not dedicated to web design, such as Microsoft Word or Quark XPress, also have the ability to function as HTML editors.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.

In biochemistry, a Ramachandran plot, originally developed in 1963 by G. N. Ramachandran, C. Ramakrishnan, and V. Sasisekharan, is a way to visualize energetically allowed regions for backbone dihedral angles ψ against φ of amino acid residues in protein structure. The figure on the left illustrates the definition of the φ and ψ backbone dihedral angles. The ω angle at the peptide bond is normally 180°, since the partial-double-bond character keeps the peptide bond planar. The figure in the top right shows the allowed φ,ψ backbone conformational regions from the Ramachandran et al. 1963 and 1968 hard-sphere calculations: full radius in solid outline, reduced radius in dashed, and relaxed tau (N-Cα-C) angle in dotted lines. Because dihedral angle values are circular and 0° is the same as 360°, the edges of the Ramachandran plot "wrap" right-to-left and bottom-to-top. For instance, the small strip of allowed values along the lower-left edge of the plot are a continuation of the large, extended-chain region at upper left.
A chemical file format is a type of data file which is used specifically for depicting molecular data. One of the most widely used is the chemical table file format, which is similar to Structure Data Format (SDF) files. They are text files that represent multiple chemical structure records and associated data fields. The XYZ file format is a simple format that usually gives the number of atoms in the first line, a comment on the second, followed by a number of lines with atomic symbols and cartesian coordinates. The Protein Data Bank Format is commonly used for proteins but is also used for other types of molecules. There are many other types which are detailed below. Various software systems are available to convert from one format to another.
Data cleansing or data cleaning is the process of detecting and correcting corrupt or inaccurate records from a record set, table, or database and refers to identifying incomplete, incorrect, inaccurate or irrelevant parts of the data and then replacing, modifying, or deleting the dirty or coarse data. Data cleansing may be performed interactively with data wrangling tools, or as batch processing through scripting or a data quality firewall.
The root mean square deviation (RMSD) or root mean square error (RMSE) is either one of two closely related and frequently used measures of the differences between true or predicted values on the one hand and observed values or an estimator on the other.

The Cambridge Structural Database (CSD) is both a repository and a validated and curated resource for the three-dimensional structural data of molecules generally containing at least carbon and hydrogen, comprising a wide range of organic, metal-organic and organometallic molecules. The specific entries are complementary to the other crystallographic databases such as the Protein Data Bank (PDB), Inorganic Crystal Structure Database and International Centre for Diffraction Data. The data, typically obtained by X-ray crystallography and less frequently by electron diffraction or neutron diffraction, and submitted by crystallographers and chemists from around the world, are freely accessible on the Internet via the CSD's parent organization's website. The CSD is overseen by the not-for-profit incorporated company called the Cambridge Crystallographic Data Centre, CCDC.
WHAT IF is a computer program used in a wide variety of computational macromolecular structure research fields. The software provides a flexible environment to display, manipulate, and analyze small and large molecules, proteins, nucleic acids, and their interactions.
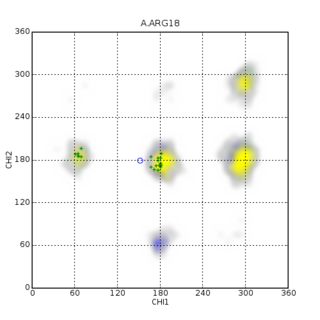
In biomolecular structure, CING stands for the Common Interface for NMR structure Generation and is known for structure and NMR data validation.
The Re-referenced Protein Chemical shift Database (RefDB) is an NMR spectroscopy database of carefully corrected or re-referenced chemical shifts, derived from the BioMagResBank (BMRB). The database was assembled by using a structure-based chemical shift calculation program to calculate expected protein (1)H, (13)C and (15)N chemical shifts from X-ray or NMR coordinate data of previously assigned proteins reported in the BMRB. The comparison is automatically performed by a program called SHIFTCOR. The RefDB database currently provides reference-corrected chemical shift data on more than 2000 assigned peptides and proteins. Data from the database indicates that nearly 25% of BMRB entries with (13)C protein assignments and 27% of BMRB entries with (15)N protein assignments require significant chemical shift reference readjustments. Additionally, nearly 40% of protein entries deposited in the BioMagResBank appear to have at least one assignment error. Users may download, search or browse the database through a number of methods available through the RefDB website. RefDB provides a standard chemical shift resource for biomolecular NMR spectroscopists, wishing to derive or compute chemical shift trends in peptides and proteins.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.
Protein Structure Evaluation Suite & Server (PROSESS) is a freely available web server for protein structure validation. It has been designed at the University of Alberta to assist with the process of evaluating and validating protein structures solved by NMR spectroscopy.
Nuclear magnetic resonance chemical shift re-referencing is a chemical analysis method for chemical shift referencing in biomolecular nuclear magnetic resonance (NMR). It has been estimated that up to 20% of 13C and up to 35% of 15N shift assignments are improperly referenced. Given that the structural and dynamic information contained within chemical shifts is often quite subtle, it is critical that protein chemical shifts be properly referenced so that these subtle differences can be detected. Fundamentally, the problem with chemical shift referencing comes from the fact that chemical shifts are relative frequency measurements rather than absolute frequency measurements. Because of the historic problems with chemical shift referencing, chemical shifts are perhaps the most precisely measurable but the least accurately measured parameters in all of NMR spectroscopy.
Protein chemical shift re-referencing is a post-assignment process of adjusting the assigned NMR chemical shifts to match IUPAC and BMRB recommended standards in protein chemical shift referencing. In NMR chemical shifts are normally referenced to an internal standard that is dissolved in the NMR sample. These internal standards include tetramethylsilane (TMS), 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) and trimethylsilyl propionate (TSP). For protein NMR spectroscopy the recommended standard is DSS, which is insensitive to pH variations. Furthermore, the DSS 1H signal may be used to indirectly reference 13C and 15N shifts using a simple ratio calculation [1]. Unfortunately, many biomolecular NMR spectroscopy labs use non-standard methods for determining the 1H, 13C or 15N “zero-point” chemical shift position. This lack of standardization makes it difficult to compare chemical shifts for the same protein between different laboratories. It also makes it difficult to use chemical shifts to properly identify or assign secondary structures or to improve their 3D structures via chemical shift refinement. Chemical shift re-referencing offers a means to correct these referencing errors and to standardize the reporting of protein chemical shifts across laboratories.
Resolution by Proxy (ResProx) is a method for assessing the equivalent X-ray resolution of NMR-derived protein structures. ResProx calculates resolution from coordinate data rather than from electron density or other experimental inputs. This makes it possible to calculate the resolution of a structure regardless of how it was solved. ResProx was originally designed to serve as a simple, single-number evaluation that allows straightforward comparison between the quality/resolution of X-ray structures and the quality of a given NMR structure. However, it can also be used to assess the reliability of an experimentally reported X-ray structure resolution, to evaluate protein structures solved by unconventional or hybrid means and to identify fraudulent structures deposited in the PDB. ResProx incorporates more than 25 different structural features to determine a single resolution-like value. ResProx values are reported in Angstroms. Tests on thousands of X-ray structures show that ResProx values match very closely to resolution values reported by X-ray crystallographers. Resolution-by-proxy values can be calculated for newly determined protein structures using a freely accessible ResProx web server. This server accepts protein coordinate data and generates a resolution estimate for that input structure.
Volume, Area, Dihedral Angle Reporter (VADAR) is a freely available protein structure validation web server that was developed as a collaboration between Dr. Brian Sykes and Dr. David Wishart at the University of Alberta. VADAR consists of over 15 different algorithms and programs for assessing and validating peptide and protein structures from their PDB coordinate data. VADAR is capable of determining secondary structure, identifying and classifying six different types of beta turns, determining and calculating the strength of C=O -- N-H hydrogen bonds, calculating residue-specific accessible surface areas (ASA), calculating residue volumes, determining backbone and side chain torsion angles, assessing local structure quality, evaluating global structure quality, and identifying residue "outliers". The results have been validated through extensive comparison to published data and careful visual inspection. VADAR produces both text and graphical output with most of the quantitative data presented in easily viewed tables. In particular, VADAR's output is presented in a vertical, tabular format with most of the sequence data, residue numbering and any other calculated property or feature presented from top to bottom, rather than from left to right.
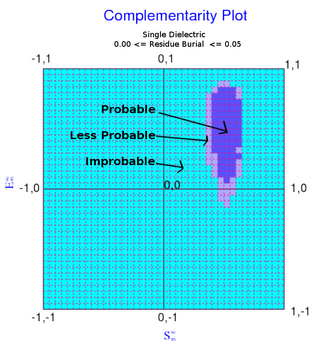
The complementarity plot (CP) is a graphical tool for structural validation of atomic models for both folded globular proteins and protein-protein interfaces. It is based on a probabilistic representation of preferred amino acid side-chain orientation, analogous to the preferred backbone orientation of Ramachandran plots). It can potentially serve to elucidate protein folding as well as binding. The upgraded versions of the software suite is available and maintained in github for both folded globular proteins as well as inter-protein complexes. The software is included in the bioinformatic tool suites OmicTools and Delphi tools.
References
- ↑ Joosten RP, te Beek TA, Krieger E, et al. (January 2011). "A series of PDB related databases for everyday needs". Nucleic Acids Res. 39 (Database issue): D411–9. doi:10.1093/nar/gkq1105. PMC 3013697 . PMID 21071423.