Related Research Articles

Systemic scleroderma, or systemic sclerosis, is an autoimmune rheumatic disease characterised by excessive production and accumulation of collagen, called fibrosis, in the skin and internal organs and by injuries to small arteries. There are two major subgroups of systemic sclerosis based on the extent of skin involvement: limited and diffuse. The limited form affects areas below, but not above, the elbows and knees with or without involvement of the face. The diffuse form also affects the skin above the elbows and knees and can also spread to the torso. Visceral organs, including the kidneys, heart, lungs, and gastrointestinal tract can also be affected by the fibrotic process. Prognosis is determined by the form of the disease and the extent of visceral involvement. Patients with limited systemic sclerosis have a better prognosis than those with the diffuse form. Death is most often caused by lung, heart, and kidney involvement. The risk of cancer is increased slightly.

Tuberous sclerosis complex (TSC) is a rare multisystem autosomal dominant genetic disease that causes non-cancerous tumours to grow in the brain and on other vital organs such as the kidneys, heart, liver, eyes, lungs and skin. A combination of symptoms may include seizures, intellectual disability, developmental delay, behavioral problems, skin abnormalities, lung disease, and kidney disease.
Neuromyelitis optica spectrum disorders (NMOSD), including neuromyelitis optica (NMO), are autoimmune diseases characterized by acute inflammation of the optic nerve and the spinal cord (myelitis). Episodes of ON and myelitis can be simultaneous or successive. A relapsing disease course is common, especially in untreated patients. In more than 80% of cases, NMO is caused by immunoglobulin G autoantibodies to aquaporin 4 (anti-AQP4), the most abundant water channel protein in the central nervous system. A subset of anti-AQP4-negative cases is associated with antibodies against myelin oligodendrocyte glycoprotein (anti-MOG). Rarely, NMO may occur in the context of other autoimmune diseases or infectious diseases. In some cases, the etiology remains unknown.

A benign tumor is a mass of cells (tumor) that does not invade neighboring tissue or metastasize. Compared to malignant (cancerous) tumors, benign tumors generally have a slower growth rate. Benign tumors have relatively well differentiated cells. They are often surrounded by an outer surface or stay contained within the epithelium. Common examples of benign tumors include moles and uterine fibroids.
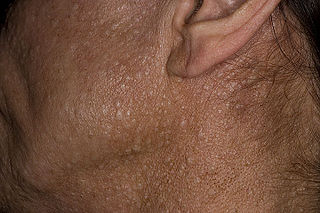
Birt–Hogg–Dubé syndrome (BHD), also Hornstein–Birt–Hogg–Dubé syndrome, Hornstein–Knickenberg syndrome, and fibrofolliculomas with trichodiscomas and acrochordons is a human, adult onset, autosomal dominant genetic disorder caused by the FLCN gene. It can cause susceptibility to kidney cancer, renal and pulmonary cysts, and noncancerous tumors of the hair follicles, called fibrofolliculomas. The symptoms seen in each family are unique, and can include any combination of the three symptoms. Fibrofolliculomas are the most common manifestation, found on the face and upper trunk in over 80% of people with BHD over the age of 40. Pulmonary cysts are equally common (84%), but only 24% of people with BHD eventually experience a collapsed lung. Kidney tumors, both cancerous and benign, occur in 14–34% of people with BHD; the associated kidney cancers are often rare hybrid tumors.

Lymphangioleiomyomatosis (LAM) is a rare, progressive and systemic disease that typically results in cystic lung destruction. It predominantly affects women, especially during childbearing years. The term sporadic LAM is used for patients with LAM not associated with tuberous sclerosis complex (TSC), while TSC-LAM refers to LAM that is associated with TSC.

Langerhans cell histiocytosis (LCH) is an abnormal clonal proliferation of Langerhans cells, abnormal cells deriving from bone marrow and capable of migrating from skin to lymph nodes.
Phakomatoses, also known neurocutaneous syndromes, are a group of multisystemic diseases that most prominently affect structures primarily derived from the ectoderm such as the central nervous system, skin and eyes. The majority of phakomatoses are single-gene disorders that may be inherited in an autosomal dominant, autosomal recessive or X-linked pattern. Presentations may vary dramatically between patients with the same particular syndrome due to mosaicism, variable expressivity, and penetrance.

Angiomyolipomas are the most common benign tumour of the kidney. Although regarded as benign, angiomyolipomas may grow such that kidney function is impaired or the blood vessels may dilate and burst, leading to bleeding.
Inflammatory demyelinating diseases (IDDs), sometimes called Idiopathic (IIDDs) due to the unknown etiology of some of them, are a heterogenous group of demyelinating diseases - conditions that cause damage to myelin, the protective sheath of nerve fibers - that occur against the background of an acute or chronic inflammatory process. IDDs share characteristics with and are often grouped together under Multiple Sclerosis. They are sometimes considered different diseases from Multiple Sclerosis, but considered by others to form a spectrum differing only in terms of chronicity, severity, and clinical course.

Baló's concentric sclerosis is a disease in which the white matter of the brain appears damaged in concentric layers, leaving the axis cylinder intact. It was described by József Mátyás Baló who initially named it "leuko-encephalitis periaxialis concentrica" from the previous definition, and it is currently considered one of the borderline forms of multiple sclerosis.

The history of tuberous sclerosis (TSC) research spans less than 200 years. TSC is a rare, multi-system genetic disease that can cause benign tumours to grow on the brain or other vital organs such as the kidneys, heart, eyes, lungs, and skin. A combination of symptoms may include seizures, developmental delay, behavioural problems and skin abnormalities, as well as lung and kidney disease. TSC is caused by mutations on either of two genes, TSC1 and TSC2, which encode for the proteins hamartin and tuberin respectively. These proteins act as tumour growth suppressors and regulate cell proliferation and differentiation. Originally regarded as a rare pathological curiosity, it is now an important focus of research into tumour formation and suppression.

Adenocarcinoma in situ (AIS) of the lung —previously included in the category of "bronchioloalveolar carcinoma" (BAC)—is a subtype of lung adenocarcinoma. It tends to arise in the distal bronchioles or alveoli and is defined by a non-invasive growth pattern. This small solitary tumor exhibits pure alveolar distribution and lacks any invasion of the surrounding normal lung. If completely removed by surgery, the prognosis is excellent with up to 100% 5-year survival.
Atypical adenomatous hyperplasia is a subtype of pneumocytic hyperplasia in the lung. It can be a precursor lesion of in situ adenocarcinoma of the lung. In prostate tissue biopsy, it can be confused for adenocarcinoma of the prostate. The needle biopsy rate is less than 1%.

Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) or Reed's syndrome is rare autosomal dominant disorder associated with benign smooth muscle tumors and an increased risk of renal cell carcinoma. It is characterised by multiple cutaneous leiomyomas and, in women, uterine leiomyomas. It predisposes for renal cell cancer, an association denominated hereditary leiomyomatosis and renal cell cancer, and it is also associated with increased risk of uterine leiomyosarcoma. The syndrome is caused by a mutation in the fumarate hydratase gene, which leads to an accumulation of fumarate. The inheritance pattern is autosomal dominant and screening can typically begin in childhood.
Atypical hemolytic uremic syndrome (aHUS), also known as complement-mediated hemolytic uremic syndrome, is an extremely rare, life-threatening, progressive disease that frequently has a genetic component. In most cases it can be effectively controlled by interruption of the complement cascade. Particular monoclonal antibodies, discussed later in the article, have proven efficacy in many cases.
Atypical adenomatous hyperplasia (AAH) is a hyperplastic lesion of the epithelial lining of pulmonary alveoli.

Multifocal micronodular pneumocyte hyperplasia (MMPH) is a subtype of pneumocytic hyperplasia.
MOG antibody disease (MOGAD) or MOG antibody-associated encephalomyelitis (MOG-EM) is an inflammatory demyelinating disease of the central nervous system. Serum anti-myelin oligodendrocyte glycoprotein antibodies are present in up to half of patients with an acquired demyelinating syndrome and have been described in association with a range of phenotypic presentations, including acute disseminated encephalomyelitis, optic neuritis, transverse myelitis, and neuromyelitis optica.
Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) is a diffuse parenchymal lung disease which often presents with symptoms of cough and shortness of breath. The pathological definition published by the World Health Organization is “a generalized proliferation of scattered single cells, small nodules, or linear proliferations of pulmonary neuroendocrine (PNE) cells that may be confined to the bronchial and bronchiolar epithelium.” The true prevalence of this disease is not known. To date, just under 200 cases have been reported in the literature. However, with an increase in recognition of this disease by radiologists and pulmonologists, the number of cases has been increasing. DIPNECH predominantly affects middle-aged women with slowly progressive lung obstruction. DIPNECH is usually discovered in one of two ways: 1) as an unexpected finding following a lung surgery; or 2) by evaluation of a patient in a pulmonary clinic with longstanding, unexplained symptoms.
References
- ↑ Behnes, C. L.; Schütze, G; Engelke, C; Bremmer, F; Gunawan, B; Radzun, H. J.; Schweyer, S (2013). "13-year-old tuberous sclerosis patient with renal cell carcinoma associated with multiple renal angiomyolipomas developing multifocal micronodular pneumocyte hyperplasia". BMC Clinical Pathology. 13: 4. doi: 10.1186/1472-6890-13-4 . PMC 3568416 . PMID 23379654.
- ↑ Ishii, M; Asano, K; Kamiishi, N; Hayashi, Y; Arai, D; Haraguchi, M; Sugiura, H; Naoki, K; Tasaka, S; Soejima, K; Sayama, K; Betsuyaku, T (2012). "Tuberous sclerosis diagnosed by incidental computed tomography findings of multifocal micronodular pneumocyte hyperplasia: A case report". Journal of Medical Case Reports. 6 (1): 352. doi: 10.1186/1752-1947-6-352 . PMC 3512476 . PMID 23072249.
- ↑ Miravet Sorribes, L; Mancheño Franch, N; Batalla Bautista, L (2013). "Multifocal micronodular pneumocyte hyperplasia in a patient with tuberous sclerosis". Archivos de Bronconeumología. 49 (1): 36–7. doi:10.1016/j.arbres.2012.06.006. PMID 22884294.
- ↑ Shintani, Y; Ohta, M; Iwasaki, T; Ikeda, N; Tomita, E; Nagano, T; Kawahara, K (2010). "A case of micronodular pneumocyte hyperplasia diagnosed through surgical resection". Annals of Thoracic and Cardiovascular Surgery. 16 (1): 45–7. PMID 20190710.
- ↑ Kobashi, Y; Sugiu, T; Mouri, K; Irei, T; Nakata, M; Oka, M (2008). "Multifocal micronodular pneumocyte hyperplasia associated with tuberous sclerosis: Differentiation from multiple atypical adenomatous hyperplasia". Japanese Journal of Clinical Oncology. 38 (6): 451–4. doi: 10.1093/jjco/hyn042 . PMID 18535095.
| | This medical sign article is a stub. You can help Wikipedia by expanding it. |