
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.
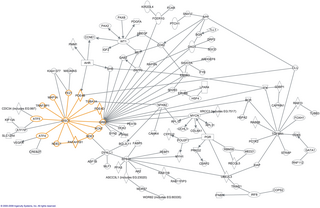
In molecular biology, an interactome is the whole set of molecular interactions in a particular cell. The term specifically refers to physical interactions among molecules but can also describe sets of indirect interactions among genes.
Protein subcellular localization prediction involves the prediction of where a protein resides in a cell, its subcellular localization.

PROSITE is a protein database. It consists of entries describing the protein families, domains and functional sites as well as amino acid patterns and profiles in them. These are manually curated by a team of the Swiss Institute of Bioinformatics and tightly integrated into Swiss-Prot protein annotation. PROSITE was created in 1988 by Amos Bairoch, who directed the group for more than 20 years. Since July 2018, the director of PROSITE and Swiss-Prot is Alan Bridge.
The cells of eukaryotic organisms are elaborately subdivided into functionally-distinct membrane-bound compartments. Some major constituents of eukaryotic cells are: extracellular space, plasma membrane, cytoplasm, nucleus, mitochondria, Golgi apparatus, endoplasmic reticulum (ER), peroxisome, vacuoles, cytoskeleton, nucleoplasm, nucleolus, nuclear matrix and ribosomes.
Human Proteinpedia, which is closely associated with Institute of Bioinformatics (IOB), Bangalore and Johns Hopkins University, is a portal for sharing and integration of human proteomic data. It allows research laboratories to contribute and maintain protein annotations. Human Protein Reference Database (HPRD) integrates data, that is deposited in Human Proteinpedia along with the existing literature curated information at the context of an individual protein. In essence, researchers can add new data to HPRD by registering to Human Proteinpedia. The data deposited in Human Proteinpedia is freely available for download. Emphasizing the importance of proteomics data disposition to public repositories, Nature Methods recommends Human Proteinpedia in their editorial. More than 70 labs participate in this effort.
Pseudo amino acid composition, or PseAAC, in molecular biology, was originally introduced by Kuo-Chen Chou in 2001 to represent protein samples for improving protein subcellular localization prediction and membrane protein type prediction. Like the vanilla amino acid composition (AAC) method, it characterizes the protein mainly using a matrix of amino-acid frequencies, which helps with dealing with proteins without significant sequential homology to other proteins. Compared to AAC, additional information are also included in the matrix to represent some local features, such as correlation between residues of a certain distance. When dealing the cases of PseAAC, the Chou's invariance theorem has been often used.

Ram Samudrala is a professor of computational biology and bioinformatics at the University at Buffalo, United States. He researches protein folding, structure, function, interaction, design, and evolution.
Edward Marcotte is a professor of biochemistry at The University of Texas at Austin, working in genetics, proteomics, and bioinformatics. Marcotte is an example of a computational biologist who also relies on experiments to validate bioinformatics-based predictions.
Computational Resources for Drug Discovery (CRDD) is an important module of the in silico module of Open Source for Drug Discovery (OSDD). The CRDD web portal provides computer resources related to drug discovery, predicting inhibitors, and predicting the ADME-Tox properties of molecules on a single platform. It caters to researchers researching computer-aided drug design by providing computational resources, and hosting a discussion forum. One of the major objectives of CRDD is to promote open source software in the field of cheminformatics and pharmacoinformatics.

PSORTdb is a database of protein subcellular localization (SCL) for bacteria and archaea. It is a member of the PSORT family of bioinformatics tools. The database consists of two datasets, ePSORTdb and cPSORTdb, which contain information determined through experimental validation and computational prediction, respectively. The ePSORTdb dataset is the largest curated collection of experimentally verified SCL data.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition/protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from those profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.
PredictProtein (PP) is an automatic service that searches up-to-date public sequence databases, creates alignments, and predicts aspects of protein structure and function. Users send a protein sequence and receive a single file with results from database comparisons and prediction methods. PP went online in 1992 at the European Molecular Biology Laboratory; since 1999 it has operated from Columbia University and in 2009 it moved to the Technische Universität München. Although many servers have implemented particular aspects, PP remains the most widely used public server for structure prediction: over 1.5 million requests from users in 104 countries have been handled; over 13000 users submitted 10 or more different queries. PP web pages are mirrored in 17 countries on 4 continents. The system is optimized to meet the demands of experimentalists not experienced in bioinformatics. This implied that we focused on incorporating only high-quality methods, and tried to collate results omitting less reliable or less important ones.
BASys is a freely available web server that can be used to perform automated, comprehensive annotation of bacterial genomes. With the advent of next generation DNA sequencing it is now possible to sequence the complete genome of a bacterium within a single day. This has led to an explosion in the number of fully sequenced microbes. In fact, as of 2013, there were more than 2700 fully sequenced bacterial genomes deposited with GenBank. However, a continuing challenge with microbial genomics is finding the resources or tools for annotating the large number of newly sequenced genomes. BASys was developed in 2005 in anticipation of these needs. In fact, BASys was the world’s first publicly accessible microbial genome annotation web server. Because of its widespread popularity, the BASys server was updated in 2011 through the addition of multiple server nodes to handle the large number of queries it was receiving.

KIAA0825 is a protein that in humans is encoded by the gene of the same name, located on chromosome 5, 5q15. It is a possible risk factor in Type II Diabetes, and associated with high levels of glucose in the blood. It is a relatively fast mutating gene, compared to other coding genes. There is however one region which is highly conserved across the species that have the gene, known as DUF4495. It is predicted to travel between the nucleus and the cytoplasm.

Transmembrane protein 179 is a protein that in humans is encoded by the TMEM179 gene. The function of transmembrane protein 179 is not yet well understood, but it is believed to have a function in the nervous system.
David S. Wishart is a Canadian researcher in metabolomics and a Distinguished University Professor in the Department of Biological Sciences and the Department of Computing Science at the University of Alberta. Wishart also holds cross appointments in the Faculty of Pharmacy and Pharmaceutical Sciences and the Department of Laboratory Medicine and Pathology in the Faculty of Medicine and Dentistry. Additionally, Wishart holds a joint appointment in metabolomics at the Pacific Northwest National Laboratory in Richland, Washington. Wishart is well known for his pioneering contributions to the fields of protein NMR spectroscopy, bioinformatics, cheminformatics and metabolomics. In 2011, Wishart founded the Metabolomics Innovation Centre (TMIC), which is Canada's national metabolomics laboratory.
FAM237A is a protein coding gene which encodes a protein of the same name. Within Homo sapiens, FAM237A is believed to be primarily expressed within the brain, with moderate heart and lesser testes expression,. FAM237A is hypothesized to act as a specific activator of receptor GPR83.