Immunodeficiency, also known as immunocompromisation, is a state in which the immune system's ability to fight infectious diseases and cancer is compromised or entirely absent. Most cases are acquired ("secondary") due to extrinsic factors that affect the patient's immune system. Examples of these extrinsic factors include HIV infection and environmental factors, such as nutrition. Immunocompromisation may also be due to genetic diseases/flaws such as SCID.
Ataxia–telangiectasia, also referred to as ataxia–telangiectasia syndrome or Louis–Bar syndrome, is a rare, neurodegenerative disease causing severe disability. Ataxia refers to poor coordination and telangiectasia to small dilated blood vessels, both of which are hallmarks of the disease. A–T affects many parts of the body:
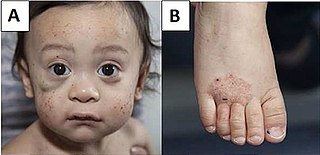
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disease characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea. It is also sometimes called the eczema-thrombocytopenia-immunodeficiency syndrome in keeping with Aldrich's original description in 1954. The WAS-related disorders of X-linked thrombocytopenia (XLT) and X-linked congenital neutropenia (XLN) may present with similar but less severe symptoms and are caused by mutations of the same gene.

ATM serine/threonine kinase or Ataxia-telangiectasia mutated, symbol ATM, is a serine/threonine protein kinase that is recruited and activated by DNA double-strand breaks, oxidative stress, topoisomerase cleavage complexes, splicing intermediates, R-loops and in some cases by single-strand DNA breaks. It phosphorylates several key proteins that initiate activation of the DNA damage checkpoint, leading to cell cycle arrest, DNA repair or apoptosis. Several of these targets, including p53, CHK2, BRCA1, NBS1 and H2AX are tumor suppressors.
Hypogammaglobulinemia is an immune system disorder in which not enough gamma globulins are produced in the blood. This results in a lower antibody count, which impairs the immune system, increasing risk of infection. Hypogammaglobulinemia may result from a variety of primary genetic immune system defects, such as common variable immunodeficiency, or it may be caused by secondary effects such as medication, blood cancer, or poor nutrition, or loss of gamma globulins in urine, as in nonselective glomerular proteinuria. Patients with hypogammaglobulinemia have reduced immune function; important considerations include avoiding use of live vaccines, and take precautionary measures when traveling to regions with endemic disease or poor sanitation such as receiving immunizations, taking antibiotics abroad, drinking only safe or boiled water, arranging appropriate medical cover in advance of travel, and ensuring continuation of any immunoglobulin infusions needed.

Nijmegen breakage syndrome (NBS) is a rare autosomal recessive congenital disorder causing chromosomal instability, probably as a result of a defect in the double Holliday junction DNA repair mechanism and/or the synthesis dependent strand annealing mechanism for repairing double strand breaks in DNA.
Primary immunodeficiencies are disorders in which part of the body's immune system is missing or does not function normally. To be considered a primary immunodeficiency (PID), the immune deficiency must be inborn, not caused by secondary factors such as other disease, drug treatment, or environmental exposure to toxins. Most primary immunodeficiencies are genetic disorders; the majority are diagnosed in children under the age of one, although milder forms may not be recognized until adulthood. While there are over 430 recognized inborn errors of immunity (IEIs) as of 2019, the vast majority of which are PIDs, most are very rare. About 1 in 500 people in the United States are born with a primary immunodeficiency. Immune deficiencies can result in persistent or recurring infections, auto-inflammatory disorders, tumors, and disorders of various organs. There are currently limited treatments available for these conditions; most are specific to a particular type of PID. Research is currently evaluating the use of stem cell transplants (HSCT) and experimental gene therapies as avenues for treatment in limited subsets of PIDs.

Neuropathy, ataxia, and retinitis pigmentosa, also known as NARP syndrome, is a rare disease with mitochondrial inheritance that causes a variety of signs and symptoms chiefly affecting the nervous system Beginning in childhood or early adulthood, most people with NARP experience numbness, tingling, or pain in the arms and legs ; muscle weakness; and problems with balance and coordination (ataxia). Many affected individuals also have vision loss caused by changes in the light-sensitive tissue that lines the back of the eye. In some cases, the vision loss results from a condition called retinitis pigmentosa. This eye disease causes the light-sensing cells of the retina gradually to deteriorate.
Purine nucleoside phosphorylase deficiency is a rare autosomal recessive metabolic disorder which results in immunodeficiency.

Non-receptor tyrosine-protein kinase TYK2 is an enzyme that in humans is encoded by the TYK2 gene.
An immune disorder is a dysfunction of the immune system. These disorders can be characterized in several different ways:

Fanconi anemia group D2 protein is a protein that in humans is encoded by the FANCD2 gene. The Fanconi anemia complementation group (FANC) currently includes FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCO, FANCP, FANCQ, FANCR, FANCS, FANCT, FANCU, FANCV, and probably FANCW. Fanconi anemia proteins, including FANCD2, are an emerging therapeutic target in cancer

Ubiquitin-conjugating enzyme E2 E3 is a protein that in humans is encoded by the UBE2E3 gene.

Ubiquitin specific peptidase 18 (USP18), also known as UBP43, is a type I interferon receptor repressor and an isopeptidase. In humans, it is encoded by the USP18 gene. USP18 is induced by the immune response to type I and III interferons, and serves as a negative regulator of type I interferon, but not type III interferon. Loss of USP18 results in increased responsiveness to type I interferons and life-threatening autoinflammatory disease in humans due to the negative regulatory function of USP18 in interferon signal transduction. Independent of this activity, USP18 is also a member of the deubiquitinating protease family of enzymes. It is known to remove ISG15 conjugates from a broad range of protein substrates, a process known as deISGylation.

T cell deficiency is a deficiency of T cells, caused by decreased function of individual T cells, it causes an immunodeficiency of cell-mediated immunity. T cells normal function is to help with the human body's immunity, they are one of the two primary types of lymphocytes.

PLAID syndrome is an inherited condition characterised by antibody deficiency and immune dysregulation, first described in 2012. The name is an acronym of "PLCG2-associated antibody deficiency and immune dysregulation". It is characterised by cold-induced urticaria, autoimmunity, atopy and humoral immune deficiency.

Immunodeficiency 26 is a rare genetic syndrome. It is characterised by absent circulating B and T cells and normal natural killer cells.

RNF144A is an E3 ubiquitin ligase belonging to the RING-between RING (RBR) family of ubiquitin ligases, whose specific members have been shown to function as RING-HECT hybrid E3 ligases. RNF144A is most closely related to RNF144B at the protein level, and the two proteins together comprise a subdomain within the RBR family of proteins. The ubiquitin ligase activity of RNF144A catalyzes ubiquitin linkages at the K6-, K11- and K48- positions of ubiquitin in vitro, and is regulated by self-association through its transmembrane domain.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
