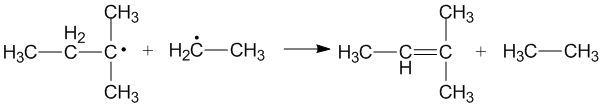Radical disproportionation encompasses a group of reactions in organic chemistry in which two radicals react to form two different non-radical products. Radicals in chemistry are defined as reactive atoms or molecules that contain an unpaired electron or electrons in an open shell. The unpaired electrons can cause radicals to be unstable and reactive. Reactions in radical chemistry can generate both radical and non-radical products. Radical disproportionation reactions can occur with many radicals in solution and in the gas phase. Due to the reactive nature of radical molecules, disproportionation proceeds rapidly and requires little to no activation energy.[1] The most thoroughly studied radical disproportionation reactions have been conducted with alkyl radicals, but there are many organic molecules that can exhibit more complex, multi-step disproportionation reactions.
In radical disproportionation reactions one molecule acts as an acceptor while the other molecule acts as a donor.[2] In the most common disproportionation reactions, a hydrogen atom is taken, or abstracted by the acceptor as the donor molecule undergoes an elimination reaction to form a double bond.[3] Other atoms such as halogens may also be abstracted during a disproportionation reaction.[4] Abstraction occurs as a head to tail reaction with the atom that is being abstracted facing the radical atom on the other molecule.
Disproportionation and steric effects
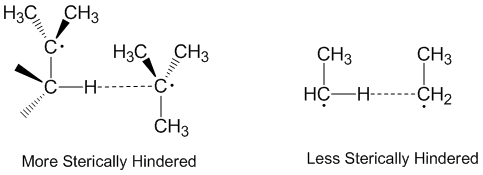
Radical disproportionation is often thought of as occurring in a linear fashion with the donor radical, the acceptor radical, and the atom being accepted all along the same axis. In fact, most disproportionation reactions do not require linear orientations in space.[2] Molecules that are more sterically hindered require arrangements that are more linear, and thus react more slowly. Steric effects play a significant role in disproportionation with ethyl radicals acting as more effective acceptors than tert-butyl radicals.[5] Tert-butyl radicals have many hydrogens on adjacent carbons to donate and steric effects often prevent tert-butyl radicals from getting close to abstracting hydrogens.[6]
Alkyl radical disproportionation
Alkyl radical disproportionation has been studied extensively in scientific literature.[6] During alkyl radical disproportionation, an alkane and an alkene are the end products and the bond order of the products increases by one over the reactants.[1] Thus the reaction is exothermic (ΔH = 50–95kcal/mol (210–400kJ/mol)) and proceeds rapidly.[6]
Cross disproportionation of alkyl radicals
Cross disproportionation occurs when two different alkyl radicals disproportionate to form two new products. Different products can be formed depending on which alkyl radical acts as a donor and which acts as an acceptor. The efficiency of primary and secondary alkyl radicals as donors depends on the steric effects and configuration of the radical acceptors.[3]
Competition with recombination
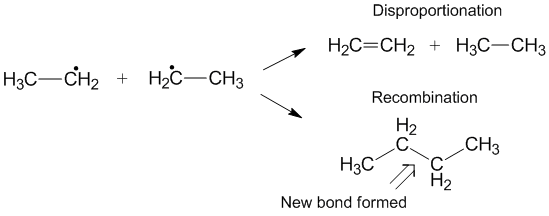
Another reaction that can sometimes occur instead of disproportionation is recombination.[6] During recombination, two radicals form one new non-radical product and one new bond. Similar to disproportionation, the recombination reaction is exothermic and requires little to no activation energy. The ratio of the rates of disproportionation to recombination is referred to as kD/kC and often favors recombination compared with disproportionation for alkyl radicals. As the number of transferable hydrogens increase, the rate constant for disproportionation increases relative to the rate constant for recombination.[3]
Kinetic isotope effect on disproportionation and recombination
When the hydrogen atoms in an alkyl radical are displaced with deuterium, disproportionation proceeds at a slightly slower rate whereas the rate of recombination remains the same. Thus disproportionation is weakly affected by the kinetic isotope effect with kH/kD = 1.20 ± 0.15 for ethylene.[7] Hydrogens and deuterons are not involved in recombination reactions. However, deuteron abstraction during disproportionation occurs more slowly than hydrogen abstraction due to the increased mass and reduced vibrational energy of deuterium, although the experimentally observed kH/kD is close to one.
Polar effects and alkoxy radical disproportionation
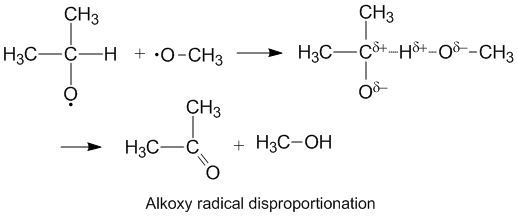
Alkoxy radicals which contain unpaired electrons on an oxygen atom display a higher kD/kC compared to alkyl radicals. The oxygen has a partial negative charge which removes electron density from the donor carbon atom thereby facilitating hydrogen abstraction. The rate of disproportionation is also aided by the more electronegative oxygen on the acceptor molecule.[6]
Termination of chain processes
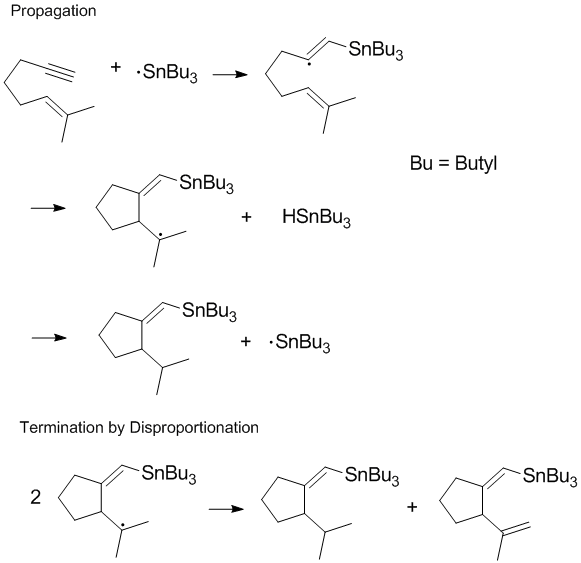
Many radical processes involve chain reactions or chain propagation with disproportionation and recombination occurring in the terminal step of the reaction.[8] Terminating chain propagation is often most significant during polymerization as the desired chain propagation cannot take place if disproportionation and recombination reactions readily occur.[8] Controlling termination products and regulating disproportionation and recombination reactions in the terminal step are important considerations in radical chemistry and polymerization. In some reactions (such as the one shown below) one or both of the termination pathways can be hindered by steric or solvent effects.[9]
Reducing disproportionation in living free radical polymerization
Many polymer chemists are concerned with limiting the rate of disproportionation during polymerization. Although disproportionation results in formation of one new double bond which may react with the polymer chain, a saturated hydrocarbon is also formed, and thus the chain reaction does not readily proceed.[10] During living free radical polymerization, termination pathways for a growing polymer chain are removed. This can be achieved through several methods, one of which is reversible termination with stable radicals. Nitroxide radicals and other stable radicals reduce recombination and disproportionation rates and control the concentration of polymeric radicals.[11]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.