Diagnosis
| | This section is empty. You can help by adding to it. (November 2021) |
| Refractory cytopenia of childhood | |
|---|---|
| Other names | RCC |
Refractory cytopenia of childhood is a subgroup of myelodysplastic syndrome (MDS), having been added to the World Health Organization classification in 2008. Before then, RCC cases were classified as childhood aplastic anemia. [1] [2] RCC is the most common form of MDS in children and adolescents, accounting for approximately half of all MDS cases. [3]
Symptoms result from underproduction of red blood cells (weakness, pallor, failure to thrive, pica), white blood cells (recurrent or overwhelming infection), and/or platelets (bleeding).[ citation needed ]
The bone marrow of patients with RCC contains islands of erythroid precursors and spare granulocytes. In some scenarios, multiple bone marrow biopsy examinations may be recommended before a diagnosis can be established.[ citation needed ]
| | This section is empty. You can help by adding to it. (November 2021) |
Bone marrow transplant is the only known curative treatment.[ citation needed ]

Anemia, also spelled anaemia and sometimes called erythrocytopenia, is a decrease in the total amount of red blood cells (RBCs) or hemoglobin in the blood or a lowered ability of the blood to carry oxygen. When anemia comes on slowly, the symptoms are often vague and may include feeling tired, weakness, shortness of breath, and a poor ability to exercise. When the anemia comes on quickly, symptoms may include confusion, lightheadedness, loss of consciousness, and increased thirst. Anemia must be significant before a person becomes noticeably pale. Additional symptoms may occur depending on the underlying cause. For people who require surgery, pre-operative anemia can increase the risk of requiring a blood transfusion following surgery.

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, so do not become healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include feeling tired, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.
Aplastic anemia is a disease in which the body fails to produce blood cells in sufficient numbers. Blood cells are produced in the bone marrow by stem cells that reside there. Aplastic anaemia causes a deficiency of all blood cell types: red blood cells, white blood cells, and platelets.
Neutropenia is an abnormally low concentration of neutrophils in the blood. Neutrophils make up the majority of circulating white blood cells and serve as the primary defense against infections by destroying bacteria, bacterial fragments and immunoglobulin-bound viruses in the blood. People with neutropenia are more susceptible to bacterial infections and, without prompt medical attention, the condition may become life-threatening.

Fanconi anaemia (FA) is a rare genetic disease resulting in impaired response to DNA damage. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), and 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity.

Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired, life-threatening disease of the blood characterized by destruction of red blood cells by the complement system, a part of the body's innate immune system. This destructive process occurs due to deficiency of the red blood cell surface protein DAF, which normally inhibits such immune reactions. Since the complement cascade attacks the red blood cells within the blood vessels of the circulatory system, the red blood cell destruction (hemolysis) is considered an intravascular hemolytic anemia. Other key features of the disease, such as the high incidence of venous blood clot formation, are incompletely understood.

Chromosome 5q deletion syndrome is an acquired, hematological disorder characterized by loss of part of the long arm of human chromosome 5 in bone marrow myelocyte cells. This chromosome abnormality is most commonly associated with the myelodysplastic syndrome.
Pancytopenia is a medical condition in which there is a reduction in the number of red and white blood cells, as well as platelets.
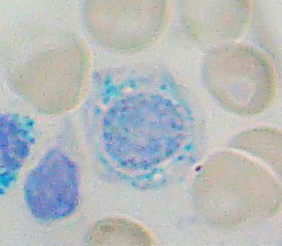
Sideroblastic anemia, or sideroachrestic anemia, is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes). In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome, which can develop into hematological malignancies.
Transient erythroblastopenia of childhood (TEC) is a slowly developing anemia of early childhood characterized by gradual onset of pallor.

Acute myeloid leukemia (AML) is a cancer of the myeloid line of blood cells, characterized by the rapid growth of abnormal cells that build up in the bone marrow and blood and interfere with normal blood cell production. Symptoms may include feeling tired, shortness of breath, easy bruising and bleeding, and increased risk of infection. Occasionally, spread may occur to the brain, skin, or gums. As an acute leukemia, AML progresses rapidly, and is typically fatal within weeks or months if left untreated.

Chronic myelomonocytic leukemia (CMML) is a type of leukemia, which are cancers of the blood-forming cells of the bone marrow. In adults, blood cells are formed in the bone marrow, by a process that is known as haematopoiesis. In CMML, there are increased numbers of monocytes and immature blood cells (blasts) in the peripheral blood and bone marrow, as well as abnormal looking cells (dysplasia) in at least one type of blood cell.
Juvenile myelomonocytic leukemia (JMML) is a serious chronic leukemia that affects children mostly aged 4 and younger. The name JMML now encompasses all diagnoses formerly referred to as juvenile chronic myeloid leukemia (JCML), chronic myelomonocytic leukemia of infancy, and infantile monosomy 7 syndrome. The average age of patients at diagnosis is 2 years old. The World Health Organization has included JMML in the category of myelodysplastic and myeloproliferative disorders.
Pearson syndrome is a mitochondrial disease characterized by sideroblastic anemia and exocrine pancreas dysfunction. Other clinical features are failure to thrive, pancreatic fibrosis with insulin-dependent diabetes and exocrine pancreatic deficiency, muscle and neurologic impairment, and, frequently, early death. It is usually fatal in infancy. The few patients who survive into adulthood often develop symptoms of Kearns–Sayre syndrome. It is caused by a deletion in mitochondrial DNA. Pearson syndrome is very rare, less than a hundred cases have been reported in medical literature worldwide.
Acute myelomonocytic leukemia (AMML) is a form of acute myeloid leukemia that involves a proliferation of CFU-GM myeloblasts and monoblasts. AMML occurs with a rapid increase amount in white blood cell count and is defined by more than 20% of myeloblast in the bone marrow. It is classified under "M4" in the French-American-British classification (FAB). It is classified under "AML, not otherwise classified" in the WHO classification.
Normocytic anemia is a type of anemia and is a common issue that occurs for men and women typically over 85 years old. Its prevalence increases with age, reaching 44 percent in men older than 85 years. The most common type of normocytic anemia is anemia of chronic disease.
Bone marrow failure occurs in individuals who produce an insufficient amount of red blood cells, white blood cells or platelets. Red blood cells transport oxygen to be distributed throughout the body’s tissue. White blood cells fight off infections that enter the body. Bone marrow also contains platelets, which trigger clotting, and thus help stop the blood flow when a wound occurs.

The Emberger syndrome is a rare, autosomal dominant, genetic disorder caused by familial or sporadic inactivating mutations in one of the two parental GATA2 genes. The mutation results in a haploinsufficiency in the levels of the gene's product, the GATA2 transcription factor. This transcription factor is critical for the embryonic development, maintenance, and functionality of blood-forming, lympathic-forming, and other tissues. The syndrome includes as its primary symptoms: serious abnormalities of the blood such as the myelodysplastic syndrome and acute myeloid leukemia; lymphedema of the lower limbs, and sensorineural hearing loss. However, the anomalies caused by GATA2 mutations are highly variable with some individuals showing little or no such symptoms even in old age while others exhibit non-malignant types of hematological anomalies; lymphedema in areas besides the lower limbs, little or no hearing loss; or anomalies in other tissues. The syndrome may present with relatively benign signs and/or symptoms and then progress rapidly or slowly to the myelodysplastic syndrome and/or acute myeloid leukemia. Alternatively, it may present with one of the latter two life-threatening disorders.
GATA2 deficiency is a grouping of several disorders caused by common defect, viz., familial or sporadic inactivating mutations in one of the two parental GATA2 genes. These autosomal dominant mutations cause a reduction, i.e. a haploinsufficiency, in the cellular levels of the gene's product, GATA2. The GATA2 protein is a transcription factor critical for the embryonic development, maintenance, and functionality of blood-forming, lymphatic-forming, and other tissue-forming stem cells. In consequence of these mutations, cellular levels of GATA2 are deficient and individuals develop over time hematological, immunological, lymphatic, or other presentations that may begin as apparently benign abnormalities but commonly progress to severe organ failure, opportunistic infections, virus infection-induced cancers, the myelodysplastic syndrome, and/or leukemia. GATA2 deficiency is a life-threatening and precancerous condition.

Transfusion-dependent anemia is a form of anemia characterized by the need for continuous blood transfusion. It is a condition that results from various diseases, and is associated with decreased survival rates. Regular transfusion is required to reduce the symptoms of anemia by increasing functional red blood cells and hemoglobin count. Symptoms may vary based on the severity of the condition and the most common symptom is fatigue. Various diseases can lead to transfusion-dependent anemia, most notably Myelodysplastic syndromes (MDS) and thalassemia. Due to the number of diseases that can cause transfusion-dependent anemia, diagnosing it is more complicated. Transfusion dependence occurs when an average of more than 2 units of blood transfused every 28 days is required over a period of at least 3 months. Myelodysplastic syndromes is often only diagnosed when patients become anemic, and transfusion-dependent thalassemia is diagnosed based on gene mutations. Screening for heterozygosity in the thalassemia gene is an option for early detection.