
Severe combined immunodeficiency (SCID), also known as Swiss-type agammaglobulinemia, is a rare genetic disorder characterized by the disturbed development of functional T cells and B cells caused by numerous genetic mutations that result in differing clinical presentations. SCID involves defective antibody response due to either direct involvement with B lymphocytes or through improper B lymphocyte activation due to non-functional T-helper cells. Consequently, both "arms" of the adaptive immune system are impaired due to a defect in one of several possible genes. SCID is the most severe form of primary immunodeficiencies, and there are now at least nine different known genes in which mutations lead to a form of SCID. It is also known as the bubble boy disease and bubble baby disease because its victims are extremely vulnerable to infectious diseases and some of them, such as David Vetter, have become famous for living in a sterile environment. SCID is the result of an immune system so highly compromised that it is considered almost absent.
Hematopoietic stem-cell transplantation (HSCT) is the transplantation of multipotent hematopoietic stem cells, usually derived from bone marrow, peripheral blood, or umbilical cord blood in order to replicate inside of a patient and to produce additional normal blood cells. It may be autologous, allogeneic or syngeneic.
Adenosine deaminase deficiency is a metabolic disorder that causes immunodeficiency. It is caused by mutations in the ADA gene. It accounts for about 10–15% of all cases of autosomal recessive forms of severe combined immunodeficiency (SCID) among non-inbred populations.

ICF syndrome is a very rare autosomal recessive immune disorder.
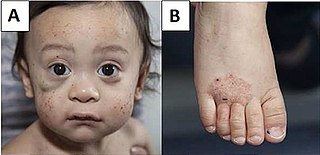
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disease characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea. It is also sometimes called the eczema-thrombocytopenia-immunodeficiency syndrome in keeping with Aldrich's original description in 1954. The WAS-related disorders of X-linked thrombocytopenia (XLT) and X-linked congenital neutropenia (XLN) may present with similar but less severe symptoms and are caused by mutations of the same gene.

Omenn syndrome is an autosomal recessive severe combined immunodeficiency. It is associated with hypomorphic missense mutations in immunologically relevant genes of T-cells such as recombination activating genes, Interleukin-7 receptor-α (IL7Rα), DCLRE1C-Artemis, RMRP-CHH, DNA-Ligase IV, common gamma chain, WHN-FOXN1, ZAP-70 and complete DiGeorge syndrome. It is fatal without treatment.

X-linked agammaglobulinemia (XLA) is a rare genetic disorder discovered in 1952 that affects the body's ability to fight infection. As the form of agammaglobulinemia that is X-linked, it is much more common in males. In people with XLA, the white blood cell formation process does not generate mature B cells, which manifests as a complete or near-complete lack of proteins called gamma globulins, including antibodies, in their bloodstream. B cells are part of the immune system and normally manufacture antibodies, which defend the body from infections by sustaining a humoral immunity response. Patients with untreated XLA are prone to develop serious and even fatal infections. A mutation occurs at the Bruton's tyrosine kinase (Btk) gene that leads to a severe block in B cell development and a reduced immunoglobulin production in the serum. Btk is particularly responsible for mediating B cell development and maturation through a signaling effect on the B cell receptor BCR. Patients typically present in early childhood with recurrent infections, in particular with extracellular, encapsulated bacteria. XLA is deemed to have a relatively low incidence of disease, with an occurrence rate of approximately 1 in 200,000 live births and a frequency of about 1 in 100,000 male newborns. It has no ethnic predisposition. XLA is treated by infusion of human antibody. Treatment with pooled gamma globulin cannot restore a functional population of B cells, but it is sufficient to reduce the severity and number of infections due to the passive immunity granted by the exogenous antibodies.
Hypogammaglobulinemia is an immune system disorder in which not enough gamma globulins are produced in the blood. This results in a lower antibody count, which impairs the immune system, increasing risk of infection. Hypogammaglobulinemia may result from a variety of primary genetic immune system defects, such as common variable immunodeficiency, or it may be caused by secondary effects such as medication, blood cancer, or poor nutrition, or loss of gamma globulins in urine, as in nonselective glomerular proteinuria. Patients with hypogammaglobulinemia have reduced immune function; important considerations include avoiding use of live vaccines, and take precautionary measures when traveling to regions with endemic disease or poor sanitation such as receiving immunizations, taking antibiotics abroad, drinking only safe or boiled water, arranging appropriate medical cover in advance of travel, and ensuring continuation of any immunoglobulin infusions needed.
Lymphoproliferative disorders (LPDs) refer to a specific class of diagnoses, comprising a group of several conditions, in which lymphocytes are produced in excessive quantities. These disorders primarily present in patients who have a compromised immune system. Due to this factor, there are instances of these conditions being equated with "immunoproliferative disorders"; although, in terms of nomenclature, lymphoproliferative disorders are a subclass of immunoproliferative disorders—along with hypergammaglobulinemia and paraproteinemias.
Enzyme replacement therapy (ERT) is a medical treatment which replaces an enzyme that is deficient or absent in the body. Usually, this is done by giving the patient an intravenous (IV) infusion of a solution containing the enzyme.
ZAP70 deficiency, or ZAP70 deficient SCID, is a rare autosomal recessive form of severe combined immunodeficiency (SCID) resulting in a lack of CD8+ T cells. People with this disease lack the capability to fight infections, and it is fatal if untreated.

X-linked severe combined immunodeficiency (X-SCID) is an immunodeficiency disorder in which the body produces very few T cells and NK cells.
Bare lymphocyte syndrome is a condition caused by mutations in certain genes of the major histocompatibility complex or involved with the processing and presentation of MHC molecules. It is a form of severe combined immunodeficiency.
Primary immunodeficiencies are disorders in which part of the body's immune system is missing or does not function normally. To be considered a primary immunodeficiency (PID), the immune deficiency must be inborn, not caused by secondary factors such as other disease, drug treatment, or environmental exposure to toxins. Most primary immunodeficiencies are genetic disorders; the majority are diagnosed in children under the age of one, although milder forms may not be recognized until adulthood. While there are over 430 recognized inborn errors of immunity (IEIs) as of 2019, the vast majority of which are PIDs, most are very rare. About 1 in 500 people in the United States are born with a primary immunodeficiency. Immune deficiencies can result in persistent or recurring infections, auto-inflammatory disorders, tumors, and disorders of various organs. There are currently limited treatments available for these conditions; most are specific to a particular type of PID. Research is currently evaluating the use of stem cell transplants (HSCT) and experimental gene therapies as avenues for treatment in limited subsets of PIDs.
The severe combined immunodeficiency (SCID) is a severe immunodeficiency genetic disorder that is characterized by the complete inability of the adaptive immune system to mount, coordinate, and sustain an appropriate immune response, usually due to absent or atypical T and B lymphocytes. In humans, SCID is colloquially known as "bubble boy" disease, as victims may require complete clinical isolation to prevent lethal infection from environmental microbes.
Hans Dieter Ochs, is an immunologist and pediatrician. He is Professor of Pediatrics, Division of Immunology, Department of Pediatrics, University of Washington School of Medicine, Seattle.
The NSG mouse is a brand of immunodeficient laboratory mice, developed and marketed by Jackson Laboratory, which carries the strain NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ. NSG branded mice are among the most immunodeficient described to date. NSG branded mice lack mature T cells, B cells, and natural killer (NK) cells. NSG branded mice are also deficient in multiple cytokine signaling pathways, and they have many defects in innate immunity. The compound immunodeficiencies in NSG branded mice permit the engraftment of a wide range of primary human cells, and enable sophisticated modeling of many areas of human biology and disease. NSG branded mice were developed in the laboratory of Dr. Leonard Shultz at Jackson Laboratory, which owns the NSG trade mark.
DOCK8 deficiency, also called DOCK8 immunodeficiency syndrome, is the autosomal recessive form of hyperimmunoglobulin E syndrome, a genetic disorder characterized by elevated immunoglobulin E levels, eosinophilia, and recurrent infections with staphylococcus and viruses. It is caused by a mutation in the DOCK8 gene.
Mice with severe combined immunodeficiency (SCIDs) are often used in the research of human disease. Human immune cells are used to develop human lymphoid organs within these immunodeficient mice, and many different types of SCID mouse models have been developed. These mice allow researchers to study the human immune system and human disease in a small animal model.

Shimon Slavin is an Israeli professor of medicine. Slavin pioneered the use of immunotherapy mediated by allogeneic donor lymphocytes and innovative methods for stem cell transplantation for the cure of hematological malignancies and solid tumors, and using hematopoietic stem cells for induction of transplantation tolerance to bone marrow and donor allografts.
