
Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

In humans, a single transverse palmar crease is a single crease that extends across the palm of the hand, formed by the fusion of the two palmar creases. Although it is found more frequently in persons with several abnormal medical conditions, it is not predictive of any of these conditions since it is also found in persons with no abnormal medical conditions. It is found in 1.5% of the world population in at least one hand.

Exophthalmos is a bulging of the eye anteriorly out of the orbit. Exophthalmos can be either bilateral or unilateral. Complete or partial dislocation from the orbit is also possible from trauma or swelling of surrounding tissue resulting from trauma.

Ectopia lentis is a displacement or malposition of the eye's lens from its normal location. A partial dislocation of a lens is termed lens subluxation or subluxated lens; a complete dislocation of a lens is termed lens luxation or luxated lens.
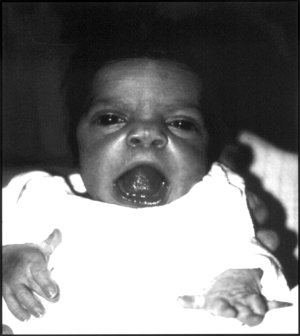
Robinow syndrome is an extremely rare genetic disorder characterized by short-limbed dwarfism, abnormalities in the head, face, and external genitalia, as well as vertebral segmentation. The disorder was first described in 1969 by human geneticist Meinhard Robinow, along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.

Larsen syndrome (LS) is a congenital disorder discovered in 1950 by Larsen and associates when they observed dislocation of the large joints and face anomalies in six of their patients. Patients with Larsen syndrome normally present with a variety of symptoms, including congenital anterior dislocation of the knees, dislocation of the hips and elbows, flattened facial appearance, prominent foreheads, and depressed nasal bridges. Larsen syndrome can also cause a variety of cardiovascular and orthopedic abnormalities. This rare disorder is caused by a genetic defect in the gene encoding filamin B, a cytoplasmic protein that is important in regulating the structure and activity of the cytoskeleton. The gene that influences the emergence of Larsen syndrome is found in chromosome region, 3p21.1-14.1, a region containing human type VII collagen gene. Larsen syndrome has recently been described as a mesenchyme disorder that affects the connective tissue of an individual. Autosomal dominant and recessive forms of the disorder have been reported, although most cases are autosomal dominant. Reports have found that in Western societies, Larsen syndrome can be found in one in every 100,000 births, but this is most likely an underestimate because the disorder is frequently unrecognized or misdiagnosed.

Micrognathism is a condition where the jaw is undersized. It is also sometimes called mandibular hypoplasia. It is common in infants, but is usually self-corrected during growth, due to the jaws' increasing in size. It may be a cause of abnormal tooth alignment and in severe cases can hamper feeding. It can also, both in adults and children, make intubation difficult, either during anesthesia or in emergency situations.

Axenfeld–Rieger syndrome is a rare autosomal dominant disorder, which affects the development of the teeth, eyes, and abdominal region.

Peters-plus syndrome or Krause–Kivlin syndrome is a hereditary syndrome defined by Peters' anomaly, dwarfism and intellectual disability.
Renal dysplasia-limb defects syndrome, also known as Ulbright–Hodes syndrome, is a very rare autosomal recessive congenital disorder. It has been described in three infants, all of whom died shortly after birth.
Woodhouse–Sakati syndrome, is a rare autosomal recessive multisystem disorder which causes malformations throughout the body, and deficiencies affecting the endocrine system.

Persistent fetal vasculature(PFV), also known as persistent fetal vasculature syndrome (PFVS), and until 1997 known primarily as persistent hyperplastic primary vitreous (PHPV), is a rare congenital anomaly which occurs when blood vessels within the developing eye, known as the embryonic hyaloid vasculature network, fail to regress as they normally would in-utero after the eye is fully developed. Defects which arise from this lack of vascular regression are diverse; as a result, the presentation, symptoms, and prognosis of affected patients vary widely, ranging from clinical insignificance to irreversible blindness. The underlying structural causes of PFV are considered to be relatively common, and the vast majority of cases do not warrant additional intervention. When symptoms do manifest, however, they are often significant, causing detrimental and irreversible visual impairment. Persistent fetal vasculature heightens the lifelong risk of glaucoma, cataracts, intraocular hemorrhages, and Retinal detachments, accounting for the visual loss of nearly 5% of the blind community in the developed world. In diagnosed cases of PFV, approximately 90% of patients with a unilateral disease have associated poor vision in the affected eye.

Lethal congenital contracture syndrome 1 (LCCS1), also called Multiple contracture syndrome, Finnish type, is an autosomal recessive genetic disorder characterized by total immobility of a fetus, detectable at around the 13th week of pregnancy. LCCS1 invariably leads to prenatal death before the 32nd gestational week. LCCS1 is one of 40 Finnish heritage diseases. It was first described in 1985 and since then, approximately 70 cases have been diagnosed.
Kapur–Toriello syndrome is a rare autosomal recessive genetic disorder. The defining feature of Kapur–Toriello syndrome is abnormal morphology of the columella, the end of the nasal septum, which in the syndrome extends below the margin of the nostrils.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.

Forkhead box protein E3 (FOXE3) also known as forkhead-related transcription factor 8 (FREAC-8) is a protein that in humans is encoded by the FOXE3 gene located on the short arm of chromosome 1.
Blepharophimosis intellectual disability syndromes are a group of rare genetic disorders which are characterized by blepharophimosis, ptosis, and intellectual disabilities. These disorders usually follow either autosomal recessive, autosomal dominant, x-linked recessive, or mitochondrial inheritance patterns.
