
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.
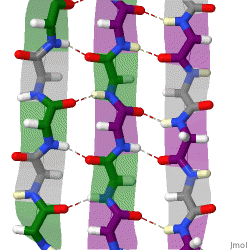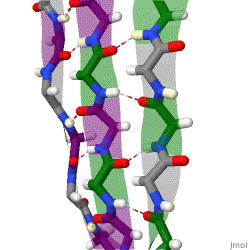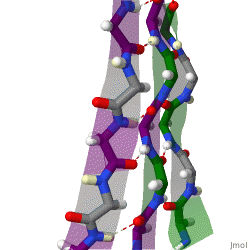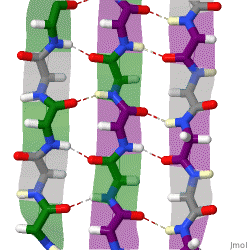
The beta sheet, (β-sheet) is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
In a chain-like biological molecule, such as a protein or nucleic acid, a structural motif is a common three-dimensional structure that appears in a variety of different, evolutionarily unrelated molecules. A structural motif does not have to be associated with a sequence motif; it can be represented by different and completely unrelated sequences in different proteins or RNA.
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.

A 310 helix is a type of secondary structure found in proteins and polypeptides. Of the numerous protein secondary structures present, the 310-helix is the fourth most common type observed; following α-helices, β-sheets and reverse turns. 310-helices constitute nearly 10–15% of all helices in protein secondary structures, and are typically observed as extensions of α-helices found at either their N- or C- termini. Because of the α-helices tendency to consistently fold and unfold, it has been proposed that the 310-helix serves as an intermediary conformation of sorts, and provides insight into the initiation of α-helix folding.
β turns are the most common form of turns—a type of non-regular secondary structure in proteins that cause a change in direction of the polypeptide chain. They are very common motifs in proteins and polypeptides. Each consists of four amino acid residues. They can be defined in two ways:
- By the possession of an intra-main-chain hydrogen bond between the CO of residue i and the NH of residue i+3;
- By having a distance of less than 7Å between the Cα atoms of residues i and i+3.
The Walker A and Walker B motifs are protein sequence motifs, known to have highly conserved three-dimensional structures. These were first reported in ATP-binding proteins by Walker and co-workers in 1982.
The Nest is a type of protein structural motif. It is a small recurring anion-binding feature of both proteins and peptides. Each consists of the main chain atoms of three consecutive amino acid residues. The main chain NH groups bind the anions while the side chain atoms are often not involved. Proline residues lack NH groups so are rare in nests. About one in 12 of amino acid residues in proteins, on average, belongs to a nest.
The Asx turn is a structural feature in proteins and polypeptides. It consists of three amino acid residues in which residue i is an aspartate (Asp) or asparagine (Asn) that forms a hydrogen bond from its sidechain CO group to the mainchain NH group of residue i+2. About 14% of Asx residues present in proteins belong to Asx turns.

Catgrips are small cation-binding molecular features of proteins and peptides. Each consists of the main chain atoms only of three consecutive amino acid residues. The first and third main chain CO groups bind the cations, often calcium, magnesium, potassium or sodium, with no side chain involvement. Many catgrips bind a water molecule instead of a cation; it is hydrogen-bonded to the first and third main chain CO groups. Catgrips are found as calcium-binding features in annexins, matrix metalloproteinases (e.g.serralysins), subtilisins and phospholipase A2. They are also observed in synthetic peptides and in cyclic hexapeptides made from alternating D,L amino acids.
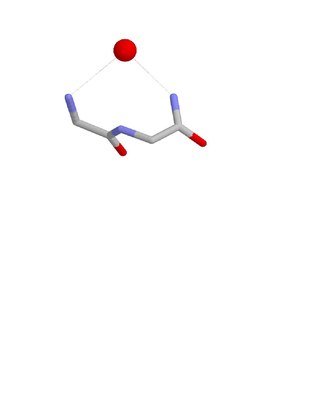
The ST turn is a structural feature in proteins and polypeptides. Each consists of three amino acid residues in which residue i is a serine (S) or threonine (T) that forms a hydrogen bond from its sidechain oxygen group to the mainchain NH group of residue i + 2.
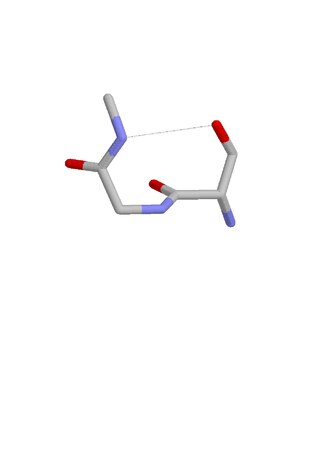
The beta bend ribbon, or beta-bend ribbon, is a structural feature in polypeptides and proteins. The shortest possible has six amino acid residues arranged as two overlapping hydrogen bonded beta turns in which the carbonyl group of residue i is hydrogen-bonded to the NH of residue i+3 while the carbonyl group of residue i+2 is hydrogen-bonded to the NH of residue i+5. In longer ribbons, this bonding is continued in peptides of 8, 10, etc., amino acid residues. A beta bend ribbon can be regarded as an aberrant 310 helix (3/10-helix) that has lost some of its hydrogen bonds. Two websites are available to facilitate finding and examining these features in proteins: Motivated Proteins; and PDBeMotif.
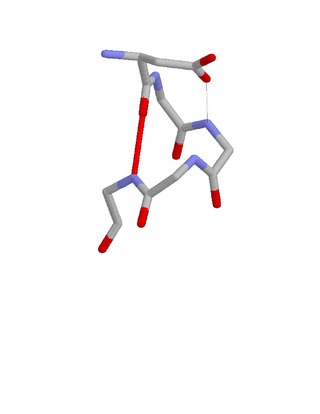
The Asx motif is a commonly occurring feature in proteins and polypeptides. It consists of four or five amino acid residues with either aspartate or asparagine as the first residue. It is defined by two internal hydrogen bonds. One is between the side chain oxygen of residue i and the main chain NH of residue i+2 or i+3; the other is between the main chain oxygen of residue i and the main chain NH of residue i+3 or i+4. Asx motifs occur commonly in proteins and polypeptides.

The ST motif is a commonly occurring feature in proteins and polypeptides. It consists of four or five amino acid residues with either serine or threonine as the first residue. It is defined by two internal hydrogen bonds. One is between the side chain oxygen of residue i and the main chain NH of residue i + 2 or i + 3; the other is between the main chain oxygen of residue i and the main chain NH of residue i + 3 or i + 4. Two websites are available for finding and examining ST motifs in proteins, Motivated Proteins: and PDBeMotif.
The term N cap describes an amino acid in a particular position within a protein or polypeptide. The N cap residue of an alpha helix is the first amino acid residue at the N terminus of the helix. More precisely, it is defined as the first residue (i) whose CO group is hydrogen-bonded to the NH group of residue i+4. Because of this it is sometimes also described as the residue prior to the helix.
The term C cap describes an amino acid in a particular position within a protein or polypeptide. The C cap residue of an alpha helix is the last amino acid residue at the C terminus of the helix. More precisely, it is defined as the last residue (i) whose NH group is hydrogen-bonded to the CO group of residue i-4. Because of this it is sometimes also described as the residue following the helix.

Beta bulge loops are commonly occurring motifs in proteins and polypeptides consisting of five to six amino acids. There are two types: type 1, which is a pentapeptide; and type 2, with six amino acids. They are regarded as a type of beta bulge, and have the alternative name of type G1 beta bulge. Compared to other beta bulges, beta bulge loops give rise to chain reversal such that they often occur at the loop ends of beta hairpins; hairpins of this sort can be described as 3:5 or 4:6. Two websites are available for finding and examining β bulge loops in proteins, Motivated Proteins: and PDBeMotif:.
The ST staple is a common four- or five-amino acid residue motif in proteins and polypeptides with serine or threonine as the C-terminal residue. It is characterized by a single hydrogen bond between the hydroxyl group of the serine or threonine and the main chain carbonyl group of residue i. Motifs are of two types, depending whether the motif has 4 or 5 residues. Most ST staples occur in conjunction with an alpha helix, and are usually associated with a slight bend in the helix. Two websites are available for finding and examining ST staples in proteins: Motivated Proteins and PDBeMotif.
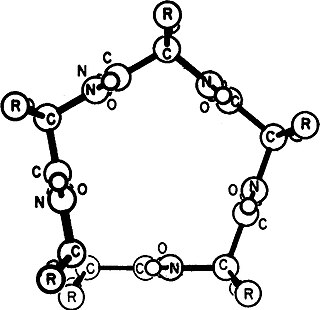
Gamma helix (or γ-helix) is a type of secondary structure in proteins that has been predicted by Pauling, Corey, and Branson, but has never been observed in natural proteins. The hydrogen bond in this type of helix was predicted to be between N-H group of one amino acid and the C=O group of the amino acid six residues earlier (or, as described by Pauling, Corey, Branson, "to the fifth amide group beyond it"). This can also be described as i + 6 → i bond and would be a continuation of the series (310 helix, alpha helix, pi helix and gamma helix). This theoretical helix contains 5.1 residues per turn.However, a fully developed gamma helix has characteristics of a structure that has 2.2 amino acid residues per turn, a rise of 2.75Å per residue, and a pseudo-cyclic (C7) structure closed by intramolecular H-bond. Depending on the amino acid's side chain (R) involved in this main-chain reversal motif, two stereoisomers can occur with their Cα-substituent located either in the axial or in the equatorial position relative to the H-bonded pseudo-cycle.

