Related Research Articles

Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.

In biochemistry, allosteric regulation is the regulation of an enzyme by binding an effector molecule at a site other than the enzyme's active site.

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.

Molecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.
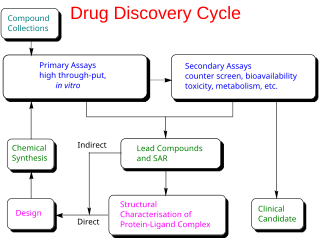
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.
Protein design is the rational design of new protein molecules to design novel activity, behavior, or purpose, and to advance basic understanding of protein function. Proteins can be designed from scratch or by making calculated variants of a known protein structure and its sequence. Rational protein design approaches make protein-sequence predictions that will fold to specific structures. These predicted sequences can then be validated experimentally through methods such as peptide synthesis, site-directed mutagenesis, or artificial gene synthesis.

Protein–protein interactions (PPIs) are physical contacts of high specificity established between two or more protein molecules as a result of biochemical events steered by interactions that include electrostatic forces, hydrogen bonding and the hydrophobic effect. Many are physical contacts with molecular associations between chains that occur in a cell or in a living organism in a specific biomolecular context.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
Macromolecular docking is the computational modelling of the quaternary structure of complexes formed by two or more interacting biological macromolecules. Protein–protein complexes are the most commonly attempted targets of such modelling, followed by protein–nucleic acid complexes.

In molecular biology, an intrinsically disordered protein (IDP) is a protein that lacks a fixed or ordered three-dimensional structure, typically in the absence of its macromolecular interaction partners, such as other proteins or RNA. IDPs range from fully unstructured to partially structured and include random coil, molten globule-like aggregates, or flexible linkers in large multi-domain proteins. They are sometimes considered as a separate class of proteins along with globular, fibrous and membrane proteins.
Protein–ligand docking is a molecular modelling technique. The goal of protein–ligand docking is to predict the position and orientation of a ligand when it is bound to a protein receptor or enzyme. Pharmaceutical research employs docking techniques for a variety of purposes, most notably in the virtual screening of large databases of available chemicals in order to select likely drug candidates. There has been rapid development in computational ability to determine protein structure with programs such as AlphaFold, and the demand for the corresponding protein-ligand docking predictions is driving implementation of software that can find accurate models. Once the protein folding can be predicted accurately along with how the ligands of various structures will bind to the protein, the ability for drug development to progress at a much faster rate becomes possible.
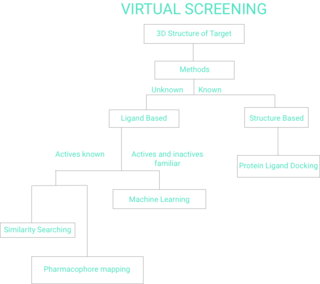
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
In the fields of computational chemistry and molecular modelling, scoring functions are mathematical functions used to approximately predict the binding affinity between two molecules after they have been docked. Most commonly one of the molecules is a small organic compound such as a drug and the second is the drug's biological target such as a protein receptor. Scoring functions have also been developed to predict the strength of intermolecular interactions between two proteins or between protein and DNA.
The program UCSF DOCK was created in the 1980s by Irwin "Tack" Kuntz's Group, and was the first docking program. DOCK uses geometric algorithms to predict the binding modes of small molecules. Brian K. Shoichet, David A. Case, and Robert C.Rizzo are codevelopers of DOCK.
Lead Finder is a computational chemistry tool designed for modeling protein-ligand interactions. This application is useful for conducting molecular docking studies and quantitatively assessing ligand binding and biological activity. Lead Finder offers free access to individual users, especially those in non-commercial and academic settings.
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design, fragment-based design, ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.
LeDock is a proprietary, flexible molecular docking software designed for the purpose of docking ligands with target proteins. It is available for Linux, macOS, and Windows.
FlexAID is a molecular docking software that can use small molecules and peptides as ligands and proteins and nucleic acids as docking targets. As the name suggests, FlexAID supports full ligand flexibility as well side-chain flexibility of the target. It does using a soft scoring function based on the complementarity of the two surfaces.
Namasivayam Gautham is a retired Professor Emeritus at the Centre of Advance Study in Crystallography and Biophysics, University of Madras. He is known for his work on DNA Crystallography, protein structure prediction and molecular docking.
References
- ↑ Halperin I; Ma B; Wolfson H; Nussinov R (June 2002). "Principles of docking: An overview of search algorithms and a guide to scoring functions". Proteins. 47 (4): 409–443. doi:10.1002/prot.10115. PMID 12001221. S2CID 7679220.
- ↑ Mustard D; Ritchie DW (August 2005). "Docking essential dynamics eigenstructures". Proteins. 60 (2): 269–274. CiteSeerX 10.1.1.134.7903 . doi:10.1002/prot.20569. PMID 15981272. S2CID 15889592.
- ↑ Shoichet BK; Stroud RM; Santi DV; Kuntz ID; Perry KM (March 1993). "Structure-based discovery of inhibitors of thymidylate synthase". Science. 259 (5100): 1445–50. Bibcode:1993Sci...259.1445S. doi:10.1126/science.8451640. PMID 8451640.
- ↑ McGann MR; Almond HR; Nicholls A; Grant JA; Brown FK (January 2003). "Gaussian docking functions". Biopolymers. 68 (1): 76–90. CiteSeerX 10.1.1.115.8784 . doi:10.1002/bip.10207. PMID 12579581.
- ↑ Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS (March 2004). "Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy". J. Med. Chem. 47 (7): 1739–1749. doi:10.1021/jm0306430. PMID 15027865.
- ↑ Jain AN (February 2003). "Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine". J. Med. Chem. 46 (4): 499–511. doi:10.1021/jm020406h. PMID 12570372.
- ↑ Zsoldos Z; Reid D; Simon A; Sadjad SB; Johnson AP (July 2007). "eHiTS: a new fast, exhaustive flexible ligand docking system". J. Mol. Graph. Model. 26 (1): 198–212. doi:10.1016/j.jmgm.2006.06.002. PMID 16860582.
- ↑ Jones G; Willett P; Glen RC; Leach AR; Taylor R (April 1997). "Development and validation of a genetic algorithm for flexible docking". J. Mol. Biol. 267 (3): 727–748. CiteSeerX 10.1.1.130.3377 . doi:10.1006/jmbi.1996.0897. PMID 9126849.
- ↑ Goodsell DS; Morris GM; Olson AJ (1996). "Automated docking of flexible ligands: applications of AutoDock". J. Mol. Recognit. 9 (1): 1–5. doi: 10.1002/(SICI)1099-1352(199601)9:1<1::AID-JMR241>3.0.CO;2-6 . PMID 8723313.