Related Research Articles
Inclusion body myositis (IBM) is the most common inflammatory muscle disease in older adults. The disease is characterized by slowly progressive weakness and wasting of both proximal muscles and distal muscles, most apparent in the finger flexors and knee extensors. IBM is often confused with an entirely different class of diseases, called hereditary inclusion body myopathies (hIBM). The "M" in hIBM is an abbreviation for "myopathy" while the "M" in IBM is for "myositis". In IBM, two processes appear to occur in the muscles in parallel, one autoimmune and the other degenerative. Inflammation is evident from the invasion of muscle fibers by immune cells. Degeneration is characterized by the appearance of holes, deposits of abnormal proteins, and filamentous inclusions in the muscle fibers. sIBM is a rare disease, with a prevalence ranging from 1 to 71 individuals per million.
Kocher–Debré–Semelaigne syndrome (KDSS) is hypothyroidism in infancy or childhood characterised by lower extremity or generalized muscular hypertrophy, myxoedema, short stature, and cognitive impairment.

Lambert–Eaton myasthenic syndrome (LEMS) is a rare autoimmune disorder characterized by muscle weakness of the limbs. It is also known as myasthenic syndrome, Eaton–Lambert syndrome, and when related to cancer, carcinomatous myopathy.
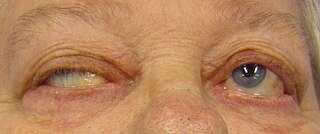
Myasthenia gravis (MG) is a long-term neuromuscular junction disease that leads to varying degrees of skeletal muscle weakness. The most commonly affected muscles are those of the eyes, face, and swallowing. It can result in double vision, drooping eyelids, and difficulties in talking and walking. Onset can be sudden. Those affected often have a large thymus or develop a thymoma.

Benign fasciculation syndrome (BFS) is characterized by fasciculation (twitching) of voluntary muscles in the body. The twitching can occur in any voluntary muscle group but is most common in the eyelids, arms, hands, fingers, legs, and feet. The tongue can also be affected. The twitching may be occasional to continuous. BFS must be distinguished from other conditions that include muscle twitches.

A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.
In medicine, myopathy is a disease of the muscle in which the muscle fibers do not function properly. Myopathy means muscle disease. This meaning implies that the primary defect is within the muscle, as opposed to the nerves or elsewhere.
Nemaline myopathy is a congenital, often hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births. It is the most common non-dystrophic myopathy.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.

MELAS is one of the family of mitochondrial diseases, which also include MIDD, MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is mitochondrial mutation, mtDNA, referred to as m.3243A>G.
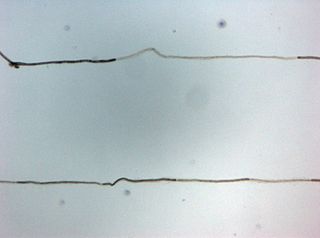
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an acquired autoimmune disease of the peripheral nervous system characterized by progressive weakness and impaired sensory function in the legs and arms. The disorder is sometimes called chronic relapsing polyneuropathy (CRP) or chronic inflammatory demyelinating polyradiculoneuropathy. CIDP is closely related to Guillain–Barré syndrome and it is considered the chronic counterpart of that acute disease. Its symptoms are also similar to progressive inflammatory neuropathy. It is one of several types of neuropathy.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.
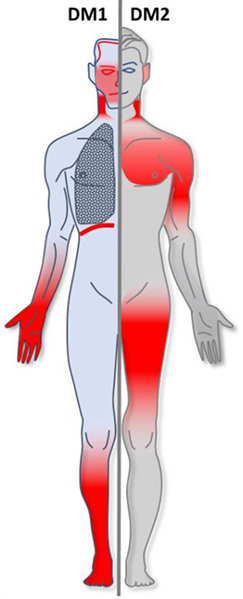
Myotonic dystrophy (DM) is a type of muscular dystrophy, a group of genetic disorders that cause progressive muscle loss and weakness. In DM, muscles are often unable to relax after contraction. Other manifestations may include cataracts, intellectual disability and heart conduction problems. In men, there may be early balding and infertility. While myotonic dystrophy can occur at any age, onset is typically in the 20s and 30s.
Congenital myopathy is a very broad term for any muscle disorder present at birth. This defect primarily affects skeletal muscle fibres and causes muscular weakness and/or hypotonia. Congenital myopathies account for one of the top neuromuscular disorders in the world today, comprising approximately 6 in 100,000 live births every year. As a whole, congenital myopathies can be broadly classified as follows:

Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of limb-girdle muscular dystrophy. There are two types of Bethlem myopathy, based on which type of collagen is affected.
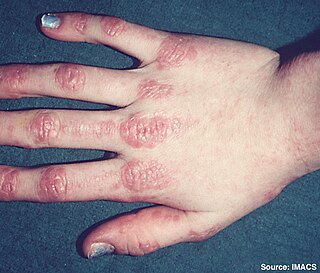
Inflammatory myopathy, also known as idiopathic inflammatory myopathy (IIM), is disease featuring muscle weakness, inflammation of muscles (myositis), and in some types, muscle pain (myalgia). The cause of much inflammatory myopathy is unknown (idiopathic), and such cases are classified according to their symptoms and signs, electromyography, MRI, and laboratory findings. It can also be associated with underlying cancer. The main classes of idiopathic inflammatory myopathy are polymyositis (PM), dermatomyositis (DM), inclusion-body myositis (IBM), immune-mediated necrotising myopathy (IMNM), and focal autoimmune myositis.
Danon disease is a metabolic disorder. Danon disease is an X-linked lysosomal and glycogen storage disorder associated with hypertrophic cardiomyopathy, skeletal muscle weakness, and intellectual disability. It is inherited in an X-linked dominant pattern.
Mitochondrially encoded tRNA phenylalanine also known as MT-TF is a transfer RNA which in humans is encoded by the mitochondrial MT-TF gene.

Camptocormia, also known as bent spine syndrome (BSS), is a symptom of a multitude of diseases that is most commonly seen in the elderly. It is identified by an abnormal thoracolumbar spinal flexion, which is a forward bending of the lower joints of the spine, occurring in a standing position. In order to be classified as BSS, the anterior flexion must be of 45 degrees anteriorly. This classification differentiates it from a similar syndrome known as kyphosis. Although camptocormia is a symptom of many diseases, there are two common origins: neurological and muscular. Camptocormia is treated by alleviating the underlying condition causing it through therapeutic measures or lifestyle changes.
Acquired non-inflammatory myopathy (ANIM) is a neuromuscular disorder primarily affecting skeletal muscle, most commonly in the limbs of humans, resulting in a weakness or dysfunction in the muscle. A myopathy refers to a problem or abnormality with the myofibrils, which compose muscle tissue. In general, non-inflammatory myopathies are a grouping of muscular diseases not induced by an autoimmune-mediated inflammatory pathway. These muscular diseases usually arise from a pathology within the muscle tissue itself rather than the nerves innervating that tissue. ANIM has a wide spectrum of causes which include drugs and toxins, nutritional imbalances, acquired metabolic dysfunctions such as an acquired defect in protein structure, and infections.
References
- ↑ Chahin N, Selcen D, Engel AG (October 2005). "Sporadic late onset nemaline myopathy". Neurology. 65 (8): 1158–64. doi:10.1212/01.wnl.0000180362.90078.dc. PMID 16148261. S2CID 23334154.
- ↑ Engel AG (November 1966). "Late-onset rod myopathy (a new syndrome?): light and electron microscopic observations in two cases". Mayo Clinic Proceedings. 41 (11): 713–41. PMID 5957590.
- ↑ Engel, W.K.; Resnick, J.S. (1966). "Late onset rod myopathy: a newly recognized, acquired, and progressive disease". Neurology. 16 (3): 308–9. doi:10.1212/wnl.16.3.299. S2CID 219213521.
- ↑ Engel WK, Oberc MA (March 1975). "Abundant nuclear rods in adult-onset rod disease". Journal of Neuropathology and Experimental Neurology. 34 (2): 119–32. doi:10.1097/00005072-197503000-00001. PMID 47386. S2CID 38570228.
- ↑ Suzuki M, Shimizu Y, Takeuchi M, Kobayashi M, Iwata M, Uchiyama S (February 2012). "Sporadic late-onset nemaline myopathy in a patient with primary Sjögren's syndrome". Journal of Neurology. 259 (2): 358–60. doi:10.1007/s00415-011-6160-4. PMID 21744311. S2CID 26449729.
- ↑ Keller CE, Hays AP, Rowland LP, Moghadaszadeh B, Beggs AH, Bhagat G (January 2006). "Adult-onset nemaline myopathy and monoclonal gammopathy". Archives of Neurology. 63 (1): 132–4. doi:10.1001/archneur.63.1.132. PMID 16401746.
- ↑ Bos MM, Overeem S, van Engelen BG, et al. (August 2006). "A case of neuromuscular mimicry". Neuromuscular Disorders. 16 (8): 510–3. doi:10.1016/j.nmd.2006.06.005. PMID 16919950. S2CID 12971698.
- ↑ Parks NE, Hanada EY (December 2012). "Maximizing functional independence in sporadic late onset nemaline myopathy". PM&R. 4 (12): 1020–3. doi:10.1016/j.pmrj.2012.06.008. PMID 23245665. S2CID 32083243.
- ↑ Milone M, Katz A, Amato AA, et al. (February 2010). "Sporadic late onset nemaline myopathy responsive to IVIg and immunotherapy". Muscle & Nerve. 41 (2): 272–6. doi:10.1002/mus.21504. PMID 19852026. S2CID 33462149.
- ↑ Hanisch F, Schneider I, Müller T, et al. (August 2013). "Behandelbarkeit der 'sporadic late onset nemaline myopathy'" [Treatability of sporadic late onset nemaline myopathy]. Der Nervenarzt (in German). 84 (8): 955–61. doi:10.1007/s00115-013-3825-5. PMID 23836301. S2CID 20819712.
- ↑ Novy J, Rosselet A, Spertini O, Lobrinus JA, Pabst T, Kuntzer T (February 2010). "Chemotherapy is successful in sporadic late onset nemaline myopathy (SLONM) with monoclonal gammopathy". Muscle & Nerve. 41 (2): 286–7. doi:10.1002/mus.21560. PMID 19918772. S2CID 36060636.