
Organometallic chemistry is the study of organometallic compounds, chemical compounds containing at least one chemical bond between a carbon atom of an organic molecule and a metal, including alkali, alkaline earth, and transition metals, and sometimes broadened to include metalloids like boron, silicon, and selenium, as well. Aside from bonds to organyl fragments or molecules, bonds to 'inorganic' carbon, like carbon monoxide, cyanide, or carbide, are generally considered to be organometallic as well. Some related compounds such as transition metal hydrides and metal phosphine complexes are often included in discussions of organometallic compounds, though strictly speaking, they are not necessarily organometallic. The related but distinct term "metalorganic compound" refers to metal-containing compounds lacking direct metal-carbon bonds but which contain organic ligands. Metal β-diketonates, alkoxides, dialkylamides, and metal phosphine complexes are representative members of this class. The field of organometallic chemistry combines aspects of traditional inorganic and organic chemistry.

Hydrogenation is a chemical reaction between molecular hydrogen (H2) and another compound or element, usually in the presence of a catalyst such as nickel, palladium or platinum. The process is commonly employed to reduce or saturate organic compounds. Hydrogenation typically constitutes the addition of pairs of hydrogen atoms to a molecule, often an alkene. Catalysts are required for the reaction to be usable; non-catalytic hydrogenation takes place only at very high temperatures. Hydrogenation reduces double and triple bonds in hydrocarbons.

The Williamson ether synthesis is an organic reaction, forming an ether from an organohalide and a deprotonated alcohol (alkoxide). This reaction was developed by Alexander Williamson in 1850. Typically it involves the reaction of an alkoxide ion with a primary alkyl halide via an SN2 reaction. This reaction is important in the history of organic chemistry because it helped prove the structure of ethers.
Supramolecular chemistry refers to the branch of chemistry concerning chemical systems composed of a discrete number of molecules. The strength of the forces responsible for spatial organization of the system range from weak intermolecular forces, electrostatic charge, or hydrogen bonding to strong covalent bonding, provided that the electronic coupling strength remains small relative to the energy parameters of the component. While traditional chemistry concentrates on the covalent bond, supramolecular chemistry examines the weaker and reversible non-covalent interactions between molecules. These forces include hydrogen bonding, metal coordination, hydrophobic forces, van der Waals forces, pi–pi interactions and electrostatic effects.

In chemistry, a supramolecular assembly is a complex of molecules held together by noncovalent bonds. While a supramolecular assembly can be simply composed of two molecules, or a defined number of stoichiometrically interacting molecules within a quaternary complex, it is more often used to denote larger complexes composed of indefinite numbers of molecules that form sphere-, rod-, or sheet-like species. Colloids, liquid crystals, biomolecular condensates, micelles, liposomes and biological membranes are examples of supramolecular assemblies, and their realm of study is known as supramolecular chemistry. The dimensions of supramolecular assemblies can range from nanometers to micrometers. Thus they allow access to nanoscale objects using a bottom-up approach in far fewer steps than a single molecule of similar dimensions.
The Ullmann reaction or Ullmann coupling, named after Fritz Ullmann, couples two aryl or alkyl groups with the help of copper. The reaction was first reported by Ullmann and his student Bielecki in 1901. It has been later shown that palladium and nickel can also be effectively used.

Macrocycles are often described as molecules and ions containing a ring of twelve or more atoms. Classical examples include the crown ethers, calixarenes, porphyrins, and cyclodextrins. Macrocycles describe a large, mature area of chemistry.

Salen refers to a tetradentate C2-symmetric ligand synthesized from salicylaldehyde (sal) and ethylenediamine (en). It may also refer to a class of compounds, which are structurally related to the classical salen ligand, primarily bis-Schiff bases. Salen ligands are notable for coordinating a wide range of different metals, which they can often stabilise in various oxidation states. For this reason salen-type compounds are used as metal deactivators. Metal salen complexes also find use as catalysts.
In chemistry, a phase-transfer catalyst or PTC is a catalyst that facilitates the transition of a reactant from one phase into another phase where reaction occurs. Phase-transfer catalysis is a special form of catalysis and can act through homogeneous catalysis or heterogeneous catalysis methods depending on the catalyst used. Ionic reactants are often soluble in an aqueous phase but insoluble in an organic phase in the absence of the phase-transfer catalyst. The catalyst functions like a detergent for solubilizing the salts into the organic phase. Phase-transfer catalysis refers to the acceleration of the reaction upon the addition of the phase-transfer catalyst.

Metal carbonyls are coordination complexes of transition metals with carbon monoxide ligands. Metal carbonyls are useful in organic synthesis and as catalysts or catalyst precursors in homogeneous catalysis, such as hydroformylation and Reppe chemistry. In the Mond process, nickel tetracarbonyl is used to produce pure nickel. In organometallic chemistry, metal carbonyls serve as precursors for the preparation of other organometallic complexes.

Dithiolene metal complexes are complexes containing 1,2-dithiolene ligands. 1,2-Dithiolene ligands, a particular case of 1,2-dichalcogenolene species along with 1,2-diselenolene derivatives, are unsaturated bidentate ligand wherein the two donor atoms are sulfur. 1,2-Dithiolene metal complexes are often referred to as "metal dithiolenes", "metallodithiolenes" or "dithiolene complexes". Most molybdenum- and tungsten-containing proteins have dithiolene-like moieties at their active sites, which feature the so-called molybdopterin cofactor bound to the Mo or W.
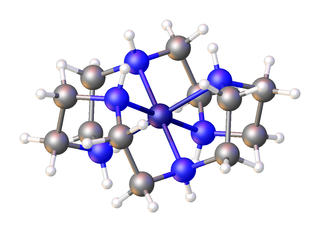
In organic chemistry, an aza-crown ether is an aza analogue of a crown ether. That is, it has a nitrogen atom in place of each oxygen atom around the ring. While the parent crown ethers have the formulae (CH2CH2O)n, the parent aza-crown ethers have the formulae (CH2CH2NH)n, where n = 3, 4, 5, 6. Well-studied aza crowns include triazacyclononane, cyclen, and hexaaza-18-crown-6.

In coordination chemistry, the first coordination sphere refers to the array of molecules and ions directly attached to the central metal atom. The second coordination sphere consists of molecules and ions that attached in various ways to the first coordination sphere.

Walter Julius Reppe was a German chemist. He is notable for his contributions to the chemistry of acetylene.

Organonickel chemistry is a branch of organometallic chemistry that deals with organic compounds featuring nickel-carbon bonds. They are used as a catalyst, as a building block in organic chemistry and in chemical vapor deposition. Organonickel compounds are also short-lived intermediates in organic reactions. The first organonickel compound was nickel tetracarbonyl Ni(CO)4, reported in 1890 and quickly applied in the Mond process for nickel purification. Organonickel complexes are prominent in numerous industrial processes including carbonylations, hydrocyanation, and the Shell higher olefin process.

A metal-phosphine complex is a coordination complex containing one or more phosphine ligands. Almost always, the phosphine is an organophosphine of the type R3P (R = alkyl, aryl). Metal phosphine complexes are useful in homogeneous catalysis. Prominent examples of metal phosphine complexes include Wilkinson's catalyst (Rh(PPh3)3Cl), Grubbs' catalyst, and tetrakis(triphenylphosphine)palladium(0).
A metal carbido complex is a coordination complex that contains a carbon atom as a ligand. They are analogous to metal nitrido complexes. Carbido complexes are a molecular subclass of carbides, which are prevalent in organometallic and inorganic chemistry. Carbido complexes represent models for intermediates in Fischer–Tropsch synthesis, olefin metathesis, and related catalytic industrial processes. Ruthenium-based carbido complexes are by far the most synthesized and characterized to date. Although, complexes containing chromium, gold, iron, nickel, molybdenum, osmium, rhenium, and tungsten cores are also known. Mixed-metal carbides are also known.
Diimines are organic compounds containing two imine (RCH=NR') groups. Common derivatives are 1,2-diketones and 1,3-diimines. These compounds are used as ligands and as precursors to heterocycles. Diimines are prepared by condensation reactions where a dialdehyde or diketone is treated with amine and water is eliminated. Similar methods are used to prepare Schiff bases and oximes.
In organic chemistry, alkynylation is an addition reaction in which a terminal alkyne is added to a carbonyl group to form an α-alkynyl alcohol.
In coordination chemistry, a macrocyclic ligand is a macrocyclic ring having at least nine atoms and three or more donor sites that serve as ligands that can bind to a central metal ion. Crown ethers and porphyrins are prominent examples. Macrocyclic ligands exhibit high affinity for metal ions.




