Related Research Articles
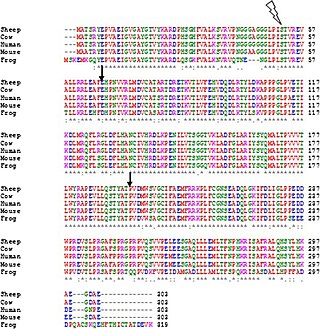
Clustal is a computer program used for multiple sequence alignment in bioinformatics. The software and its algorithms have gone through several iterations, with ClustalΩ (Omega) being the latest version as of 2011. It is available as standalone software, via a web interface, and through a server hosted by the European Bioinformatics Institute.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff.
Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. The latest version of Pfam, 37.0, was released in June 2024 and contains 21,979 families. It is currently provided through InterPro website.

Multiple sequence alignment (MSA) is the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. These alignments are used to infer evolutionary relationships via phylogenetic analysis and can highlight homologous features between sequences. Alignments highlight mutation events such as point mutations, insertion mutations and deletion mutations, and alignments are used to assess sequence conservation and infer the presence and activity of protein domains, tertiary structures, secondary structures, and individual amino acids or nucleotides.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
T-Coffee is a multiple sequence alignment software using a progressive approach. It generates a library of pairwise alignments to guide the multiple sequence alignment. It can also combine multiple sequences alignments obtained previously and in the latest versions can use structural information from Protein Data Bank (PDB) files (3D-Coffee). It has advanced features to evaluate the quality of the alignments and some capacity for identifying occurrence of motifs (Mocca). It produces alignment in the aln format (Clustal) by default, but can also produce PIR, MSF, and FASTA format. The most common input formats are supported.
MAVID is a multiple sequence alignment program suitable for the alignment of large numbers of DNA sequences. The sequences can be small mitochondrial genomes or large genomic regions up to megabases long. The latest version is 2.0.4.
Protein subfamily is a level of protein classification, based on their close evolutionary relationship. It is below the larger levels of protein superfamily and protein family.
28S ribosomal RNA is the structural ribosomal RNA (rRNA) for the large subunit (LSU) of eukaryotic cytoplasmic ribosomes, and thus one of the basic components of all eukaryotic cells. It has a size of 25S in plants and 28S in mammals, hence the alias of 25S–28S rRNA.
The Eukaryotic Linear Motif (ELM) resource is a computational biology resource for investigating short linear motifs (SLiMs) in eukaryotic proteins. It is currently the largest collection of linear motif classes with annotated and experimentally validated linear motif instances.
SUPERFAMILY is a database and search platform of structural and functional annotation for all proteins and genomes. It classifies amino acid sequences into known structural domains, especially into SCOP superfamilies. Domains are functional, structural, and evolutionary units that form proteins. Domains of common Ancestry are grouped into superfamilies. The domains and domain superfamilies are defined and described in SCOP. Superfamilies are groups of proteins which have structural evidence to support a common evolutionary ancestor but may not have detectable sequence homology.

Dehydrogenase/reductase member 7B is an enzyme encoded by the DHRS7B gene in humans, found on chromosome 17p11.2. DHRS7B encodes a protein that is predicted to function in steroid hormone regulation. A deletion in the chromosomal region 17p11.2 has been associated with Smith-Magenis Syndrome, a genetic developmental disorder.

UPF0172 protein FAM158A, also known as c14orf122 or CGI112, is a protein that in humans is encoded by the FAM158A gene located on chromosome 14q11.2.
The European Nucleotide Archive (ENA) is a repository providing free and unrestricted access to annotated DNA and RNA sequences. It also stores complementary information such as experimental procedures, details of sequence assembly and other metadata related to sequencing projects. The archive is composed of three main databases: the Sequence Read Archive, the Trace Archive and the EMBL Nucleotide Sequence Database. The ENA is produced and maintained by the European Bioinformatics Institute and is a member of the International Nucleotide Sequence Database Collaboration (INSDC) along with the DNA Data Bank of Japan and GenBank.

DEP Domain Containing Protein 1B also known as XTP1, XTP8, HBV XAg-Transactivated Protein 8, [formerly referred to as BRCC3] is a human protein encoded by a gene of similar name located on chromosome 5.
EPD is a biological database and web resource of eukaryotic RNA polymerase II promoters with experimentally defined transcription start sites. Originally, EPD was a manually curated resource relying on transcript mapping experiments targeted at individual genes and published in academic journals. More recently, automatically generated promoter collections derived from electronically distributed high-throughput data produced with the CAGE or TSS-Seq protocols were added as part of a special subsection named EPDnew. The EPD web server offers additional services, including an entry viewer which enables users to explore the genomic context of a promoter in a UCSC Genome Browser window, and direct links for uploading EPD-derived promoter subsets to associated web-based promoter analysis tools of the Signal Search Analysis (SSA) and ChIP-Seq servers. EPD also features a collection of position weight matrices (PWMs) for common promoter sequence motifs.

Alexander George Bateman is a computational biologist and Head of Protein Sequence Resources at the European Bioinformatics Institute (EBI), part of the European Molecular Biology Laboratory (EMBL) in Cambridge, UK. He has led the development of the Pfam biological database and introduced the Rfam database of RNA families. He has also been involved in the use of Wikipedia for community-based annotation of biological databases.

Desmond Gerard Higgins is a Professor of Bioinformatics at University College Dublin, widely known for CLUSTAL, a series of computer programs for performing multiple sequence alignment. According to Nature, Higgins' papers describing CLUSTAL are among the top ten most highly cited scientific papers of all time.
Marilyn S. Kozak is an American professor of biochemistry at the Robert Wood Johnson Medical School. She was previously at the University of Medicine and Dentistry of New Jersey before the school was merged. She was awarded a PhD in microbiology by Johns Hopkins University studying the synthesis of the Bacteriophage MS2, advised by Daniel Nathans.

Proline-rich protein 30 is a protein in humans that is encoded for by the PRR30 gene. PRR30 is a member in the family of Proline-rich proteins characterized by their intrinsic lack of structure. Copy number variations in the PRR30 gene have been associated with an increased risk for neurofibromatosis.
References
- 1 2 Thompson, Julie D.; Higgins, Desmond G.; Gibson, Toby J. (1994). "CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice". Nucleic Acids Research. 22 (22): 4673–4680. doi:10.1093/nar/22.22.4673. ISSN 0305-1048. PMC 308517 . PMID 7984417.
- 1 2 3 Toby Gibson publications indexed by Google Scholar
- ↑ Toby Gibson publications from Europe PubMed Central
- 1 2 Thompson, J. (1997). "The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools". Nucleic Acids Research. 25 (24): 4876–4882. doi:10.1093/nar/25.24.4876. ISSN 1362-4962. PMC 147148 . PMID 9396791.
- ↑ Thompson, J. D.; Higgins, D. G.; Gibson, T. J. (1994). "CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice". Nucleic Acids Research. 22 (22): 4673–4680. doi:10.1093/nar/22.22.4673. PMC 308517 . PMID 7984417.
- ↑ Van Noorden, R.; Maher, B.; Nuzzo, R. (2014). "The top 100 papers: Nature explores the most-cited research of all time". Nature . 514 (7524): 550–3. doi: 10.1038/514550a . PMID 25355343.
- 1 2 3 Anon (2019). "Toby (James) Gibson: EMBL, Heidelberg, Germany". uni-halle.de. Martin Luther University of Halle-Wittenberg. Archived from the original on 2019-07-05.
- ↑ Gibson, Toby James (1984). Studies on the Epstein-Barr virus genome. cam.ac.uk (PhD thesis). University of Cambridge. OCLC 499859334. EThOS uk.bl.ethos.352786.
- ↑ Kumar, Manjeet; Gouw, Marc; Michael, Sushama; Sámano-Sánchez, Hugo; Pancsa, Rita; Glavina, Juliana; Diakogianni, Athina; Valverde, Jesús Alvarado; Bukirova, Dayana; Čalyševa, Jelena; Palopoli, Nicolas; Davey, Norman E; Chemes, Lucía B; Gibson, Toby J (2019). "ELM—the eukaryotic linear motif resource in 2020". Nucleic Acids Research. doi: 10.1093/nar/gkz1030 . ISSN 0305-1048. PMC 7145657 . PMID 31680160.
| International | |
|---|---|
| National | |
| Academics | |