
Polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it to a large enough amount to study in detail. PCR was invented in 1983 by the American biochemist Kary Mullis at Cetus Corporation; Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. DNA polymerase adds nucleotides after binding to the RNA primer and synthesis the whole strand. Later, the RNA strands must be removed accurately and replace them with DNA nucleotides forming a gap region known as a nick that is filled in using an enzyme called ligase. The removal process of the RNA primer requires several enzymes, such as Fen1, Lig1, and others that work in coordination with DNA polymerase, to ensure the removal of the RNA nucleotides and the addition of DNA nucleotides. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable. Primers can be designed in laboratory for specific reactions such as polymerase chain reaction (PCR). When designing PCR primers, there are specific measures that must be taken into consideration, like the melting temperature of the primers and the annealing temperature of the reaction itself. Moreover, the DNA binding sequence of the primer in vitro has to be specifically chosen, which is done using a method called basic local alignment search tool (BLAST) that scans the DNA and finds specific and unique regions for the primer to bind.
Protein engineering is the process of developing useful or valuable proteins. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It is also a product and services market, with an estimated value of $168 billion by 2017.
Helicase-dependent amplification (HDA) is a method for in vitro DNA amplification that takes place at a constant temperature.
Random amplification of polymorphic DNA (RAPD), pronounced "rapid", is a type of polymerase chain reaction (PCR), but the segments of DNA that are amplified are random. The scientist performing RAPD creates several arbitrary, short primers, then proceeds with the PCR using a large template of genomic DNA, hoping that fragments will amplify. By resolving the resulting patterns, a semi-unique profile can be gleaned from an RAPD reaction.
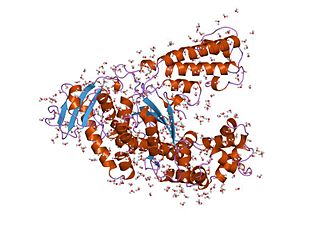
Taq polymerase is a thermostable DNA polymerase I named after the thermophilic eubacterial microorganism Thermus aquaticus, from which it was originally isolated by Chien et al. in 1976. Its name is often abbreviated to Taq or Taq pol. It is frequently used in the polymerase chain reaction (PCR), a method for greatly amplifying the quantity of short segments of DNA.
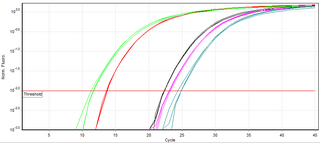
A real-time polymerase chain reaction is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, not at its end, as in conventional PCR. Real-time PCR can be used quantitatively and semi-quantitatively.
Nested polymerase chain reaction is a modification of polymerase chain reaction intended to reduce non-specific binding in products due to the amplification of unexpected primer binding sites.
The overlap extension polymerase chain reaction is a variant of PCR. It is also referred to as Splicing by overlap extension / Splicing by overhang extension (SOE) PCR. It is used to insert specific mutations at specific points in a sequence or to splice smaller DNA fragments into a larger polynucleotide.
TaqMan probes are hydrolysis probes that are designed to increase the specificity of quantitative PCR. The method was first reported in 1991 by researcher Kary Mullis at Cetus Corporation, and the technology was subsequently developed by Hoffmann-La Roche for diagnostic assays and by Applied Biosystems for research applications.
Nucleic acid sequence-based amplification, commonly referred to as NASBA, is a method in molecular biology which is used to produce multiple copies of single stranded RNA. NASBA is a two-step process that takes RNA and anneals specially designed primers, then utilizes an enzyme cocktail to amplify it.
The polymerase chain reaction (PCR) is a commonly used molecular biology tool for amplifying DNA, and various techniques for PCR optimization which have been developed by molecular biologists to improve PCR performance and minimize failure.
Polymerase cycling assembly is a method for the assembly of large DNA oligonucleotides from shorter fragments. The process uses the same technology as PCR, but takes advantage of DNA hybridization and annealing as well as DNA polymerase to amplify a complete sequence of DNA in a precise order based on the single stranded oligonucleotides used in the process. It thus allows for the production of synthetic genes and even entire synthetic genomes.
The versatility of polymerase chain reaction (PCR) has led to a large number of variants of PCR.
Multiple displacement amplification (MDA) is a DNA amplification technique. This method can rapidly amplify minute amounts of DNA samples to a reasonable quantity for genomic analysis. The reaction starts by annealing random hexamer primers to the template: DNA synthesis is carried out by a high fidelity enzyme, preferentially Φ29 DNA polymerase. Compared with conventional PCR amplification techniques, MDA does not employ sequence-specific primers but amplifies all DNA, generates larger-sized products with a lower error frequency, and works at a constant temperature. MDA has been actively used in whole genome amplification (WGA) and is a promising method for application to single cell genome sequencing and sequencing-based genetic studies.
A primer dimer (PD) is a potential by-product in the polymerase chain reaction (PCR), a common biotechnological method. As its name implies, a PD consists of two primer molecules that have attached (hybridized) to each other because of strings of complementary bases in the primers. As a result, the DNA polymerase amplifies the PD, leading to competition for PCR reagents, thus potentially inhibiting amplification of the DNA sequence targeted for PCR amplification. In quantitative PCR, PDs may interfere with accurate quantification.
COLD-PCR is a modified polymerase chain reaction (PCR) protocol that enriches variant alleles from a mixture of wildtype and mutation-containing DNA. The ability to preferentially amplify and identify minority alleles and low-level somatic DNA mutations in the presence of excess wildtype alleles is useful for the detection of mutations. Detection of mutations is important in the case of early cancer detection from tissue biopsies and body fluids such as blood plasma or serum, assessment of residual disease after surgery or chemotherapy, disease staging and molecular profiling for prognosis or tailoring therapy to individual patients, and monitoring of therapy outcome and cancer remission or relapse. Common PCR will amplify both the major (wildtype) and minor (mutant) alleles with the same efficiency, occluding the ability to easily detect the presence of low-level mutations. The capacity to detect a mutation in a mixture of variant/wildtype DNA is valuable because this mixture of variant DNAs can occur when provided with a heterogeneous sample – as is often the case with cancer biopsies. Currently, traditional PCR is used in tandem with a number of different downstream assays for genotyping or the detection of somatic mutations. These can include the use of amplified DNA for RFLP analysis, MALDI-TOF genotyping, or direct sequencing for detection of mutations by Sanger sequencing or pyrosequencing. Replacing traditional PCR with COLD-PCR for these downstream assays will increase the reliability in detecting mutations from mixed samples, including tumors and body fluids.
Hot start PCR is a modified form of conventional polymerase chain reaction (PCR) that reduces the presence of undesired products and primer dimers due to non-specific DNA amplification at room temperatures. Because the results of PCR are so useful, many variations and modifications of the procedure were developed in order to achieve a higher yields, hot start PCR is one of them. Hot start PCR follows the same principles as the conventional PCR - in that it uses DNA polymerase to synthesise DNA from a single stranded template, however, it utilises additional heating and separation methods, such as inactivating or inhibiting the binding of Taq polymerase and late addition of Taq polymerase, to increase product yield as well as provide a higher specificity and sensitivity. Non-specific binding and priming or formation of primer dimers are minimized by completing the reaction mix after denaturation. Some ways to complete reaction mixes at high temperatures involve modifications that block DNA polymerase activity in low temperatures, use of modified deoxyribonucleotide triphosphates (dNTPs), and the physical addition of one of the essential reagents after denaturation. The results of this procedure has many applications both medically and industrially. For example, applications of PCR including forensics, paternity testing, biodefence, cloning, mutation detection, genetic testing and DNA sequencing.
Multiple Annealing and Looping Based Amplification Cycles (MALBAC) is a quasilinear whole genome amplification method. Unlike conventional DNA amplification methods that are non-linear or exponential, MALBAC utilizes special primers that allow amplicons to have complementary ends and therefore to loop, preventing DNA from being copied exponentially. This results in amplification of only the original genomic DNA and therefore reduces amplification bias. MALBAC is “used to create overlapped shotgun amplicons covering most of the genome”. For next generation sequencing, MALBAC is followed by regular PCR which is used to further amplify amplicons.

RNase H-dependent PCR (rhPCR) is a modification of the standard PCR technique. In rhPCR, the primers are designed with a removable amplification block on the 3’ end. Amplification of the blocked primer is dependent on the cleavage activity of a hyperthermophilic archaeal Type II RNase H enzyme during hybridization to the complementary target sequence. This RNase H enzyme possesses several useful characteristics that enhance the PCR. First, it has very little enzymatic activity at low temperature, enabling a “hot start PCR” without modifications to the DNA polymerase. Second, the cleavage efficiency of the enzyme is reduced in the presence of mismatches near the RNA residue. This allows for reduced primer dimer formation, detection of alternative splicing variants, ability to perform multiplex PCR with higher numbers of PCR primers, and the ability to detect single-nucleotide polymorphisms.






