The SN1 reaction is a substitution reaction in organic chemistry, the name of which refers to the Hughes-Ingold symbol of the mechanism. "SN" stands for "nucleophilic substitution", and the "1" says that the rate-determining step is unimolecular. Thus, the rate equation is often shown as having first-order dependence on the substrate and zero-order dependence on the nucleophile. This relationship holds for situations where the amount of nucleophile is much greater than that of the intermediate. Instead, the rate equation may be more accurately described using steady-state kinetics. The reaction involves a carbocation intermediate and is commonly seen in reactions of secondary or tertiary alkyl halides under strongly basic conditions or, under strongly acidic conditions, with secondary or tertiary alcohols. With primary and secondary alkyl halides, the alternative SN2 reaction occurs. In inorganic chemistry, the SN1 reaction is often known as the dissociative substitution. This dissociation pathway is well-described by the cis effect. A reaction mechanism was first proposed by Christopher Ingold et al. in 1940. This reaction does not depend much on the strength of the nucleophile, unlike the SN2 mechanism. This type of mechanism involves two steps. The first step is the ionization of alkyl halide in the presence of aqueous acetone or ethyl alcohol. This step provides a carbocation as an intermediate.
In organic chemistry, Markovnikov's rule or Markownikoff's rule describes the outcome of some addition reactions. The rule was formulated by Russian chemist Vladimir Markovnikov in 1870.
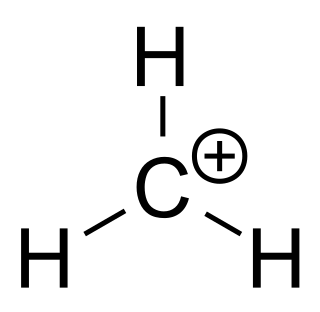
A carbocation is an ion with a positively charged carbon atom. Among the simplest examples are the methenium CH+
3, methanium CH+
5 and vinyl C
2H+
3 cations. Occasionally, carbocations that bear more than one positively charged carbon atom are also encountered.

The Beckmann rearrangement, named after the German chemist Ernst Otto Beckmann (1853–1923), is a rearrangement of an oxime functional group to substituted amides. The rearrangement has also been successfully performed on haloimines and nitrones. Cyclic oximes and haloimines yield lactams.
The Friedel–Crafts reactions are a set of reactions developed by Charles Friedel and James Crafts in 1877 to attach substituents to an aromatic ring. Friedel–Crafts reactions are of two main types: alkylation reactions and acylation reactions. Both proceed by electrophilic aromatic substitution.
A sigmatropic reaction in organic chemistry is a pericyclic reaction wherein the net result is one σ-bond is changed to another σ-bond in an uncatalyzed intramolecular reaction. The name sigmatropic is the result of a compounding of the long-established sigma designation from single carbon–carbon bonds and the Greek word tropos, meaning turn. In this type of rearrangement reaction, a substituent moves from one part of a π-bonded system to another part in an intramolecular reaction with simultaneous rearrangement of the π system. True sigmatropic reactions are usually uncatalyzed, although Lewis acid catalysis is possible. Sigmatropic reactions often have transition-metal catalysts that form intermediates in analogous reactions. The most well-known of the sigmatropic rearrangements are the [3,3] Cope rearrangement, Claisen rearrangement, Carroll rearrangement, and the Fischer indole synthesis.
In chemistry, a dehydration reaction is a chemical reaction that involves the loss of water from the reacting molecule or ion. Dehydration reactions are common processes, the reverse of a hydration reaction.
The Hofmann rearrangement is the organic reaction of a primary amide to a primary amine with one fewer carbon atom. The reaction involves oxidation of the nitrogen followed by rearrangement of the carbonyl and nitrogen to give an isocyanate intermediate. The reaction can form a wide range of products, including alkyl and aryl amines.

2-Butanol, or sec-butanol, is an organic compound with formula CH3CH(OH)CH2CH3. Its structural isomers are 1-butanol. isobutanol, and tert-butanol. 2-Butanol is chiral and thus can be obtained as either of two stereoisomers designated as (R)-(−)-2-butanol and (S)-(+)-2-butanol. It is normally encountered as a 1:1 mixture of the two stereoisomers — a racemic mixture.
A 1,2-rearrangement or 1,2-migration or 1,2-shift or Whitmore 1,2-shift is an organic reaction where a substituent moves from one atom to another atom in a chemical compound. In a 1,2 shift the movement involves two adjacent atoms but moves over larger distances are possible. In the example below the substituent R moves from carbon atom C2 to C3.
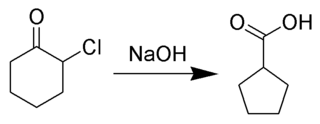
The Favorskii rearrangement is principally a rearrangement of cyclopropanones and α-halo ketones that leads to carboxylic acid derivatives. In the case of cyclic α-halo ketones, the Favorskii rearrangement constitutes a ring contraction. This rearrangement takes place in the presence of a base, sometimes hydroxide, to yield a carboxylic acid but most of the time either an alkoxide base or an amine to yield an ester or an amide, respectively. α,α'-Dihaloketones eliminate HX under the reaction conditions to give α,β-unsaturated carbonyl compounds.
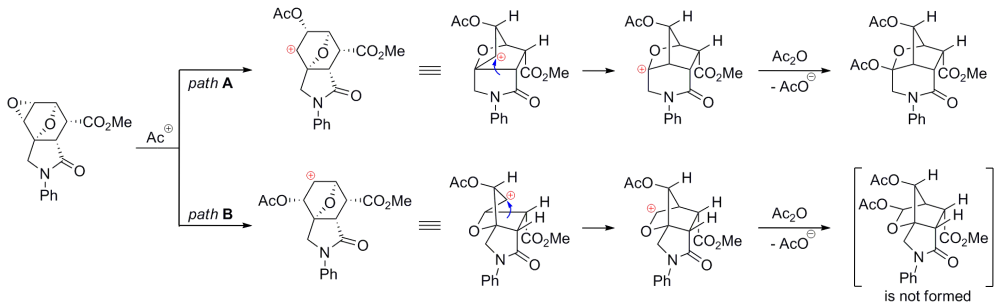
In organic chemistry, neighbouring group participation has been defined by the International Union of Pure and Applied Chemistry (IUPAC) as the interaction of a reaction centre with a lone pair of electrons in an atom or the electrons present in a pi bond contained within the parent molecule but not conjugated with the reaction centre. When NGP is in operation it is normal for the reaction rate to be increased. It is also possible for the stereochemistry of the reaction to be abnormal when compared with a normal reaction. While it is possible for neighbouring groups to influence many reactions in organic chemistry this page is limited to neighbouring group effects seen with carbocations and SN2 reactions.
In organic chemistry, the term 2-norbornyl cation describes one of the three carbocations formed from derivatives of norbornane. Though 1-norbornyl and 7-norbornyl cations have been studied, the most extensive studies and vigorous debates have been centered on the exact structure of the 2-norbornyl cation.
The Demjanov rearrangement is the chemical reaction of primary amines with nitrous acid to give rearranged alcohols. It involves substitution by a hydroxyl group with a possible ring expansion. It is named after the Russian chemist Nikolai Jakovlevich Demjanov (1861–1938).
The Hammett equation in organic chemistry describes a linear free-energy relationship relating reaction rates and equilibrium constants for many reactions involving benzoic acid derivatives with meta- and para-substituents to each other with just two parameters: a substituent constant and a reaction constant. This equation was developed and published by Louis Plack Hammett in 1937 as a follow-up to qualitative observations in a 1935 publication.
The Kornblum–DeLaMare rearrangement is a rearrangement reaction in organic chemistry in which a primary or secondary organic peroxide is converted to the corresponding ketone and alcohol under acid or base catalysis. The reaction is relevant as a tool in organic synthesis and is a key step in the biosynthesis of prostaglandins.
The Wallach rearrangement, also named Wallach transformation, is a name reaction in the organic chemistry. It is named after Otto Wallach, who discovered this reaction in 1880. In general it is a strong acid-promoted conversion of azoxybenzenes into hydroxyazobenzenes.

The Westphalen–Lettré rearrangement is a classic organic reaction in organic chemistry describing a rearrangement reaction of cholestane-3β,5α,6β-triol diacetate with acetic anhydride and sulfuric acid. In this reaction one equivalent of water is lost, a double bond is formed at C10–C11 and importantly the methyl group at the C10 position migrates to the C5 position.

Hans Meerwein was a German chemist. Several reactions and reagents bear his name, most notably the Meerwein–Ponndorf–Verley reduction, the Wagner–Meerwein rearrangement, the Meerwein arylation reaction, and Meerwein's salt.
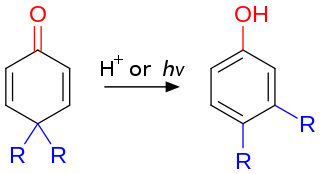
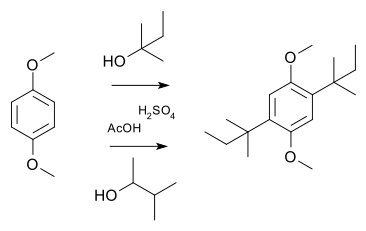
The dienone–phenol rearrangement is a reaction in organic chemistry first reported in 1921 by Karl von Auwers and Karl Ziegler. A common example of dienone–phenol rearrangement is 4,4-disubstituted cyclohexadienone converting into a stable 3,4-disubstituted phenol in presence of acid. A similar rearrangement is possible with a 2,2-disubstituted cyclohexadienone to its corresponding disubstituted phenol. Usually this type of rearrangement is spontaneous unless a dichloromethyl group is present at the 4th position or the process is otherwise blocked.