Tumors of the hematopoietic and lymphoid tissues or tumours of the haematopoietic and lymphoid tissues are tumors that affect the blood, bone marrow, lymph, and lymphatic system. Because these tissues are all intimately connected through both the circulatory system and the immune system, a disease affecting one will often affect the others as well, making aplasia, myeloproliferation and lymphoproliferation closely related and often overlapping problems. While uncommon in solid tumors, chromosomal translocations are a common cause of these diseases. This commonly leads to a different approach in diagnosis and treatment of hematological malignancies. Hematological malignancies are malignant neoplasms ("cancer"), and they are generally treated by specialists in hematology and/or oncology. In some centers "hematology/oncology" is a single subspecialty of internal medicine while in others they are considered separate divisions. Not all hematological disorders are malignant ("cancerous"); these other blood conditions may also be managed by a hematologist.

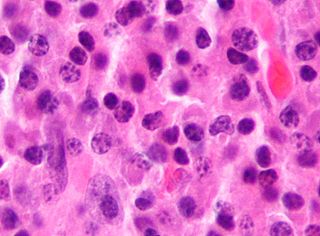
Anaplastic large-cell lymphoma (ALCL) refers to a group of non-Hodgkin lymphomas in which aberrant T cells proliferate uncontrollably. Considered as a single entity, ALCL is the most common type of peripheral lymphoma and represents ~10% of all peripheral lymphomas in children. The incidence of ALCL is estimated to be 0.25 cases per 100,000 people in the United States of America. There are four distinct types of anaplastic large-cell lymphomas that on microscopic examination share certain key histopathological features and tumor marker proteins. However, the four types have very different clinical presentations, gene abnormalities, prognoses, and/or treatments.

A myeloid sarcoma is a solid tumor composed of immature white blood cells called myeloblasts. A chloroma is an extramedullary manifestation of acute myeloid leukemia; in other words, it is a solid collection of leukemic cells occurring outside of the bone marrow.
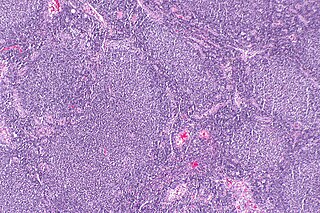
Follicular lymphoma (FL) is a cancer that involves certain types of white blood cells known as lymphocytes. The cancer originates from the uncontrolled division of specific types of B-cells known as centrocytes and centroblasts. These cells normally occupy the follicles in the germinal centers of lymphoid tissues such as lymph nodes. The cancerous cells in FL typically form follicular or follicle-like structures in the tissues they invade. These structures are usually the dominant histological feature of this cancer.
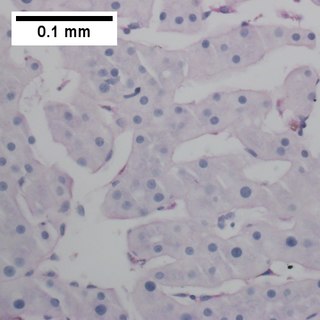
Primary effusion lymphoma (PEL) is classified as a diffuse large B cell lymphoma. It is a rare malignancy of plasmablastic cells that occurs in individuals that are infected with the Kaposi's sarcoma-associated herpesvirus. Plasmablasts are immature plasma cells, i.e. lymphocytes of the B-cell type that have differentiated into plasmablasts but because of their malignant nature do not differentiate into mature plasma cells but rather proliferate excessively and thereby cause life-threatening disease. In PEL, the proliferating plasmablastoid cells commonly accumulate within body cavities to produce effusions, primarily in the pleural, pericardial, or peritoneal cavities, without forming a contiguous tumor mass. In rare cases of these cavitary forms of PEL, the effusions develop in joints, the epidural space surrounding the brain and spinal cord, and underneath the capsule which forms around breast implants. Less frequently, individuals present with extracavitary primary effusion lymphomas, i.e., solid tumor masses not accompanied by effusions. The extracavitary tumors may develop in lymph nodes, bone, bone marrow, the gastrointestinal tract, skin, spleen, liver, lungs, central nervous system, testes, paranasal sinuses, muscle, and, rarely, inside the vasculature and sinuses of lymph nodes. As their disease progresses, however, individuals with the classical effusion-form of PEL may develop extracavitary tumors and individuals with extracavitary PEL may develop cavitary effusions.

Acute myeloid leukemia (AML) is a cancer of the myeloid line of blood cells, characterized by the rapid growth of abnormal cells that build up in the bone marrow and blood and interfere with normal blood cell production. Symptoms may include feeling tired, shortness of breath, easy bruising and bleeding, and increased risk of infection. Occasionally, spread may occur to the brain, skin, or gums. As an acute leukemia, AML progresses rapidly, and is typically fatal within weeks or months if left untreated.
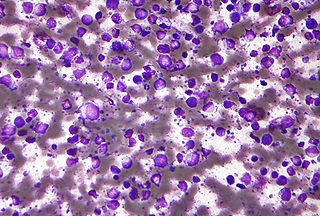
Diffuse large B-cell lymphoma (DLBCL) is a cancer of B cells, a type of lymphocyte that is responsible for producing antibodies. It is the most common form of non-Hodgkin lymphoma among adults, with an annual incidence of 7–8 cases per 100,000 people per year in the US and UK. This cancer occurs primarily in older individuals, with a median age of diagnosis at ~70 years, although it can occur in young adults and, in rare cases, children. DLBCL can arise in virtually any part of the body and, depending on various factors, is often a very aggressive malignancy. The first sign of this illness is typically the observation of a rapidly growing mass or tissue infiltration that is sometimes associated with systemic B symptoms, e.g. fever, weight loss, and night sweats.
Plasmacytoid dendritic cells (pDCs) are a rare type of immune cell that are known to secrete large quantities of type 1 interferon (IFNs) in response to a viral infection. They circulate in the blood and are found in peripheral lymphoid organs. They develop from bone marrow hematopoietic stem cells and constitute < 0.4% of peripheral blood mononuclear cells (PBMC). Other than conducting antiviral mechanisms, pDCs are considered to be key in linking the innate and adaptive immune systems. However, pDCs are also responsible for participating in and exacerbating certain autoimmune diseases like lupus. pDCs that undergo malignant transformation cause a rare hematologic disorder, blastic plasmacytoid dendritic cell neoplasm.

ETV6 protein is a transcription factor that in humans is encoded by the ETV6 gene. The ETV6 protein regulates the development and growth of diverse cell types, particularly those of hematological tissues. However, its gene, ETV6 frequently suffers various mutations that lead to an array of potentially lethal cancers, i.e., ETV6 is a clinically significant proto-oncogene in that it can fuse with other genes to drive the development and/or progression of certain cancers. However, ETV6 is also an anti-oncogene or tumor suppressor gene in that mutations in it that encode for a truncated and therefore inactive protein are also associated with certain types of cancers.
Richter's transformation (RT), also known as Richter's syndrome, is the conversion of chronic lymphocytic leukemia (CLL) or its variant, small lymphocytic lymphoma (SLL), into a new and more aggressively malignant disease. CLL is the circulation of malignant B lymphocytes with or without the infiltration of these cells into lymphatic or other tissues while SLL is the infiltration of these malignant B lymphocytes into lymphatic and/or other tissues with little or no circulation of these cells in the blood. CLL along with its SLL variant are grouped together in the term CLL/SLL.

Platelet-derived growth factor receptor beta is a protein that in humans is encoded by the PDGFRB gene. Mutations in PDGFRB are mainly associated with the clonal eosinophilia class of malignancies.

Acute megakaryoblastic leukemia (AMKL) is life-threatening leukemia in which malignant megakaryoblasts proliferate abnormally and injure various tissues. Megakaryoblasts are the most immature precursor cells in a platelet-forming lineage; they mature to promegakaryocytes and, ultimately, megakaryocytes which cells shed membrane-enclosed particles, i.e. platelets, into the circulation. Platelets are critical for the normal clotting of blood. While malignant megakaryoblasts usually are the predominant proliferating and tissue-damaging cells, their similarly malignant descendants, promegakaryocytes and megakaryocytes, are variable contributors to the malignancy.

Marginal zone B-cell lymphomas, also known as marginal zone lymphomas (MZLs), are a heterogeneous group of lymphomas that derive from the malignant transformation of marginal zone B-cells. Marginal zone B cells are innate lymphoid cells that normally function by rapidly mounting IgM antibody immune responses to antigens such as those presented by infectious agents and damaged tissues. They are lymphocytes of the B-cell line that originate and mature in secondary lymphoid follicles and then move to the marginal zones of mucosa-associated lymphoid tissue, the spleen, or lymph nodes. Mucosa-associated lymphoid tissue is a diffuse system of small concentrations of lymphoid tissue found in various submucosal membrane sites of the body such as the gastrointestinal tract, mouth, nasal cavity, pharynx, thyroid gland, breast, lung, salivary glands, eye, skin and the human spleen.

Extranodal NK/T-cell lymphoma, nasal type (ENKTCL-NT) is a rare type of lymphoma that commonly involves midline areas of the nasal cavity, oral cavity, and/or pharynx At these sites, the disease often takes the form of massive, necrotic, and extremely disfiguring lesions. However, ENKTCL-NT can also involve the eye, larynx, lung, gastrointestinal tract, skin, and various other tissues. ENKTCL-NT mainly affects adults; it is relatively common in Asia and to lesser extents Mexico, Central America, and South America but is rare in Europe and North America. In Korea, ENKTCL-NT often involves the skin and is reported to be the most common form of cutaneous lymphoma after mycosis fungoides.
Myeloid leukemia is a type of leukemia affecting myeloid tissue.
Clonal hypereosinophilia, also termed primary hypereosinophilia or clonal eosinophilia, is a grouping of hematological disorders all of which are characterized by the development and growth of a pre-malignant or malignant population of eosinophils, a type of white blood cell that occupies the bone marrow, blood, and other tissues. This population consists of a clone of eosinophils, i.e. a group of genetically identical eosinophils derived from a sufficiently mutated ancestor cell.
GATA2 deficiency is a grouping of several disorders caused by common defect, namely, familial or sporadic inactivating mutations in one of the two parental GATA2 genes. Being the gene haploinsufficient, mutations that cause a reduction in the cellular levels of the gene's product, GATA2, are autosomal dominant. The GATA2 protein is a transcription factor critical for the embryonic development, maintenance, and functionality of blood-forming, lymphatic-forming, and other tissue-forming stem cells. In consequence of these mutations, cellular levels of GATA2 are deficient and individuals develop over time hematological, immunological, lymphatic, or other presentations that may begin as apparently benign abnormalities but commonly progress to severe organ failure, opportunistic infections, virus infection-induced cancers, the myelodysplastic syndrome, and/or leukemia. GATA2 deficiency is a life-threatening and precancerous condition.
Epstein–Barr virus–associated lymphoproliferative diseases are a group of disorders in which one or more types of lymphoid cells, i.e. B cells, T cells, NK cells, and histiocytic-dendritic cells, are infected with the Epstein–Barr virus (EBV). This causes the infected cells to divide excessively, and is associated with the development of various non-cancerous, pre-cancerous, and cancerous lymphoproliferative disorders (LPDs). These LPDs include the well-known disorder occurring during the initial infection with the EBV, infectious mononucleosis, and the large number of subsequent disorders that may occur thereafter. The virus is usually involved in the development and/or progression of these LPDs although in some cases it may be an "innocent" bystander, i.e. present in, but not contributing to, the disease.
Monomorphic epitheliotropic intestinal T cell lymphoma (MEITL) is an extremely rare peripheral T-cell lymphoma that involves the malignant proliferation of a type of lymphocyte, the T cell, in the gastrointestinal tract. Over time, these T cells commonly spread throughout the mucosal lining of a portion of the GI tract, lead to GI tract nodules and ulcerations, and cause symptoms such as abdominal pain, weight loss, diarrhea, obstruction, bleeding, and/or perforation.
T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) is a malignancy of B cells. B-cells are lymphocytes that normally function in the humoral immunity component of the adaptive immune system by secreting antibodies that, for example, bind to and neutralize invasive pathogens. Among the various forms of B-cell lymphomas, THRLBCL is a rarely occurring subtype of the diffuse large B-cell lymphomas (DLBCL). DLBCL are a large group of lymphomas that account for ~25% of all non-Hodgkin lymphomas worldwide. THRLBCL is distinguished from the other DLBCL subtypes by the predominance of non-malignant T-cell lymphocytes and histiocytes over malignant B-cells in its tumors and tissue infiltrates.
