Congenital adrenal hyperplasia due to 21-hydroxylase deficiency
Other names
21-OH CAH
CT scan shows enlarged adrenals with masses consistent with congenital adrenal hyperplasia due to 21-hydroxylase deficiency (image credit: NICHD/A. Mallappa, D. Merke)
1:18,000 to 1:14,000 (classical forms); 1:1000 to 1:50 (nonclassical forms)
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency (CAH) is a genetic disorder characterized by impaired production of cortisol in the adrenal glands.[1]
It is classified as an inherited metabolic disorder. CAH is an autosomal recessive condition since it results from inheriting two copies of the faulty CYP21A2 gene responsible for 21-hydroxylase enzyme deficiency. The most common forms of CAH are: classical form, usually diagnosed at birth, and nonclassical, late onset form, typically diagnosed during childhood or adolescence, although it can also be identified in adulthood when seeking medical help for fertility concerns or other related issues, such as PCOS or menstrual irregularities.[1] Carriers for the alleles of the nonclassical forms may have no syptoms, such form of CAH is sometimes called cryptic form.[2][3] Congenital adrenal hyperplasia due to 21-hydroxylase deficiency in all its forms accounts for over 95% of diagnosed cases of all types of congenital adrenal hyperplasia.[4] Unless another specific enzyme is mentioned, CAH in most contexts refers to 21-hydroxylase deficiency, and different mutations related to enzyme impairment have been mapped on protein structures of the enzyme.[5] It is one of the most common autosomal recessive genetic diseases in humans.[6][7][8]
Due to loss of 21-hydroxylase function, patients are unable to efficiently synthesize cortisol. As a result, ACTH levels increase, leading to adrenocortical hyperplasia and overproduction of cortisol precursors, which are used in the synthesis of sex steroids, which can lead to signs of androgen excess, including ambiguous genitalia in newborn girls and rapid postnatal growth in both sexes.[1] In severe cases of CAH in females, surgical reconstruction may be considered to create more female-appearing external genitalia. However, there is ongoing debate regarding timing and necessity of surgery. The way CAH affects the organism is complicated, and not everyone who has it will show signs or have symptoms.[4] Individuals with CAH may face challenges related to growth impairment during childhood and fertility issues during adulthood. Psychosocial aspects such as gender identity development and mental health should also be taken into consideration when managing individuals with CAH.[1] Overall prognosis for individuals with appropriate medical care is good; however, lifelong management under specialized care is required to ensure optimal outcomes.[1]
Treatment for CAH involves hormone replacement therapy to provide adequate levels of glucocorticoids and mineralocorticoids. Regular monitoring is necessary to optimize hormone balance and minimize potential complications associated with long-term glucocorticoid exposure.[1]
Presentation
CAH can occur in various forms. The clinical presentation of each form is different and depends to a large extent on the kind of the underlying 21-hydroxylase enzyme defect.[9] Classical forms appear in infancy, and nonclassical forms appear in late childhood. The presentation in patients with classical CAH can be further subdivided into two forms: salt-wasting and simple-virilizing, depending on whether mineralocorticoid deficiency presents or absents, respectively.[1][10][11] This subtyping is often not clinically meaningful, because all patients with classical form lose salt to some degree, and clinical presentations may overlap.[12]
Severe, early onset 21-hydroxylase deficient CAH
The two most serious neonatal consequences of 21-hydroxylase deficiency are the following: life-threatening salt-wasting crises in the first month of life (for male and female infants alike) and severe virilization of female infants which can cause genital ambiguity.[13][14] The subdivision of the early onset CAH into salt-wasting and simple-virilizing forms, which is based on the capacity of the adrenal to produce small amounts of aldosterone in the simple-virilizing form, is often not clinically meaningful, because clinical presentations overlap and all patients lose salt to some degree.[12]
Salt-wasting crises in infancy
Excessive levels of androgens of adrenal origin[15][16][17][18] have little effect on the genitals of male infants with classical CAH.[15] If a male infant with CAH goes undetected during newborn screening, he will exhibit normal health and physical appearance upon examination, leading to his timely discharge from the hospital.[19][20][21][22]
Aldosterone insufficiency in salt-wasting CAH results in significant loss of sodium in the urine. Urinary sodium concentrations may exceed 50 mEq/L. Due to the loss of salt at that rate, the infant is unable to maintain proper blood volume and begins to suffer from dehydration due to hyponatremia by the end of the first week of life. Potassium and acid excretion are also impaired when mineralocorticoid activity is deficient, and hyperkalemia and metabolic acidosis gradually develop. When there's not enough cortisol in the body due to the classical CAH, it becomes harder to have proper blood circulation. Inadequate circulation can cause problems like spitting and difficulty gaining weight in babies. Furthermore, infants with salt-wasting CAH may experience even worse symptoms like vomiting, extreme dehydration, and their circulation can suddenly fail (which is called circulatory shock) within two or three weeks after birth.[19]
Upon admission to a medical facility, the infant between the ages of 1 and 3 weeks will exhibit signs of both underweight and dehydration.[19] The blood pressure may be abnormally low. Basic laboratory analyses will indicate low levels of sodium (hyponatremia), typically falling between 105 and 125 mEq/L Na+ in serum samples. These infants may also experience severe hyperkalemia with potassium (K+) levels exceeding 10 mEq/L, along with significant metabolic acidosis. Hypoglycemia (low blood sugar) might also be observed. This collection of manifestations is referred to as a "salt-wasting crisis" which poses an imminent risk of fatal consequences if left untreated.[23]
Despite the severity of this condition, affected infants respond swiftly to treatment involving hydrocortisone administration along with intravenous infusion of saline solution and dextrose supplementation. Consequently, normal blood volume is promptly restored alongside stabilization of blood pressure and restoration of appropriate body sodium levels, leading to reversal of hyperkalemia. With prompt and apt management measures in place, most infants are no longer at risk within a span of approximately 24 hours.[24][25][26]
Virilization of female infants
The androgen backdoor pathway (red arrows) roundabout testosterone embedded in within conventional androgen synthesis that lead to 5α-dihydrotestosterone through testosterone
CAH is a genetic disorder characterized by impaired production of cortisol in the adrenal glands.[1][4]
Production of cortisol begins at week 8 of fetal life.[30][31][32]
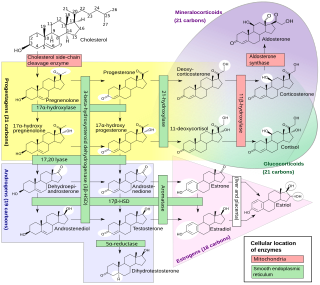
The 21-hydroxylase enzyme is essential in conversion of progesterone and 17OHP into 11-deoxycorticosterone and 11-deoxycortisol, respectively.[4][33] This process is done through hydroxylation at C-21 position.[34] It was described in at least 1953 that impaired steroid hydroxylation at C-21 position happens in congenital adrenal hyperplasia and is accompanied by excessive amounts of 17OHP that leads to virilism.[35]
In the insufficiency of 21-hydroxylase to participate in the biosynthesis of cortisol, the 21-hydroxylation in the zona fasciculata of the adrenal cortex is impaired, so 17OHP and progesterone will not be properly converted into 11-deoxycortisol and 11-deoxycorticosterone, respectively − the precursors for cortisol and aldosterone. As the plasma concentration of cortisol and aldosterone decreases, ACTH levels increase, leading to excessive production and accumulation of cortisol precursors (especially 17OHP), which are eventually transferred to androsterone that is a feedstock for other androgens.[36] Other androgens may be additionally produced from 17OHP, due to its elevated levels, that leads, inter alia, to its 5α-reduction.[37] These additional androgens are produced via the so-called "backdoor pathway" (see red arrows on the diagram).[38][39] For example, in this "backdoor" pathway, 5α-dihydrotestosterone is produced with roundabout of testosterone as an intermediate product.[37][40][41] Some of the androgens produced by the backdoor pathway are those that cannot be converted to estrogens by aromatase, causing prenatal virilization,[42] and making them the dominant androgens in classic 21-hydroxylase deficiency.[8][27]
Virilization of genetically female (XX) infants usually produces obvious genital ambiguity. Inside the pelvis, the ovaries are normal and since they have not been exposed to testicular anti-Müllerian hormone (AMH), the uterus, fallopian tubes, upper vagina, and other Müllerian structures are normally formed as well. However, the high levels of testosterone[43] in the blood can enlarge the phallus, partially or completely close the vaginal opening, enclose the urethral groove so that it opens at the base of the phallus, on the shaft or even at the tip like a boy. Testosterone can cause the labial skin to become as thin and rugate as a scrotum,[44][45] but cannot produce palpable gonads (i.e., testes) in the folds.[46][47][48][49][50][51]
Thus, depending on the severity of hyperandrogenism, a female infant can be mildly affected, obviously ambiguous, or so severely virilized as to appear to be a male. Andrea Prader devised the following Prader scale as a way of describing the degree of virilization.[52]
An infant at stage 1 has a mildly large clitoris and slightly reduced vaginal opening size. This degree may go unnoticed or may be simply assumed to be within normal variation.
Stages 2 and 3 represent progressively more severe degrees of virilization. The genitalia are obviously abnormal to the eye, with a phallus intermediate in size and a small vaginal opening.
Stage 4 looks more male than female, with an empty scrotum and a phallus the size of a normal penis, but not quite free enough of the perineum to be pulled onto the abdomen toward the umbilicus (i.e., what is termed a chordee in a male). The single small urethral/vaginal opening at the base or on the shaft of the phallus would be considered a hypospadias in a male. X-rays taken after dye injection into this opening reveal the internal connection with the upper vagina and uterus. This common opening can predispose to urinary obstruction and infection.
Stage 5 denotes complete male virilization, with a normally formed penis with the urethral opening at or near the tip. The scrotum is normally formed but empty. The internal pelvic organs include normal ovaries and uterus, and the vagina connects internally with the urethra as in Stage 4. These infants are not visibly ambiguous, and are usually assumed to be ordinary boys with undescended testes. In most cases, the diagnosis of CAH is not suspected until signs of salt-wasting develop a week later.[53][54][55][56]
When the genitalia are determined to be ambiguous at birth, CAH is one of the leading diagnostic possibilities. Evaluation reveals the presence of a uterus, extreme elevation of 17OHP,[57] levels of testosterone[43] approaching or exceeding the male range[58] but low AMH levels. The karyotype is that of an ordinary female: 46,XX. With this information, the diagnosis of CAH is readily made and female sex confirmed.[53][59][60][61][62]
Evaluation of ambiguous genitalia is described in detail elsewhere. In most cases it is possible to confirm and assign female sex within 12–36 hours of birth. The exception are the rare, completely virilized genetic females (Prader stage 5), who present the most challenging assignment and surgery dilemmas.[63][64]
When the degree of ambiguity is obvious, corrective surgery is usually offered and performed, but reconstructive surgery on infant genitalia has controversies.[65][66][67]
Infertility observed in adult males with classica CAH has been associated with testicular adrenal rest tumors (TART) that may originate during childhood.[68] Delayed diagnosis of CAH was associated with a higher risk of developing TART, which may be attributable to poor disease control early in life,[69] emphasizing the need of early detection and management of CAH, and, once CAH is detected, detection and management of TART.[70] TART in prepubertal males with classical CAH could be found during childhood (20%). Martinez-Aguayo et al. reported differences in markers of gonadal function in a subgroup of patients, especially in those with inadequate control.[71] Early glucocorticoid therapy may be beneficial to the reduction of TART.[72] Early detection and management of TART in CAH patients is important for preserving testicular function and preventing long-term complications that may impact fertility,[70] To avoid long-term complications in male patients with CAH, regular ultrasound examination of the scrotum is recommended,[70] especially in patients presenting with the most profound phenotypes of CAH and suboptimal endocrine regulation of the condition.[73] Subsequently, male subjects with nonclassic CAH rarely present with TARTs, and even though these tumors do not affect fertility and do not require regular ultrasound examination of the scrotum.[74]
Female fertility
Women with classical CAH have statistically reduced fertility, especially those with the salt-losing form.[75] Live birth rate is 33–50% in simple virilized form of CAH, and 0–10% in most severe salt-wasting form. In the nonclassical form of CAH the live birth is 63–90%, similar to the age-matched control groups.[76]
Sex assignment issues and controversies
Classical CAH leads to female pseudohermaphroditism at birth, and is the most common case of sex ambiguity, the second one is mixed gonadal dysgenesis. Most commonly, at birth, the phallus enlarges, so it is larger than normal female but smaller than normal male. Instead of separate urethral and vaginal openings, there is an urogenital sinus that is often covered by tissue resulting from the posterior fusion of the labioscrotal ridges. Therefore, different degrees of external genital abnormalities can be found, ranging from normal perineum to penile urethra.[76]
There are no difficulties assigning appropriate sex for most infants with CAH. Genetic males have normal male genitalia and gonads and simply need hormone replacement. Most virilized females are assigned and raised as girls even if their genitalia are ambiguous or look more male than female. They have normal ovaries and uterus and potential fertility with hormone replacement and surgery. The complex issues encountered in appropriate sex assignment for severely virilized XX infants have contributed to a deeper understanding of gender identity and sexual orientation. Consequently, this matter remains an ongoing subject of debate within medical discourse.[77][78][79]
Until the 1950s, some virilized XX infants were assigned and raised as girls, and some as boys. Most developed gender identities congruent with their sex of rearing. In a few cases of male rearing, a sex reassignment was attempted in mid-childhood when newly discovered karyotyping revealed "female" chromosomes. These reassignments were rarely successful, leading New Zealand American psychologist John Money and other influential psychologists and physicians to conclude that gender identity was (1) unrelated to chromosomes, (2) primarily a result of social learning, and (3) could not be easily changed after infancy.[77][78]
By the 1960s, CAH was well understood, karyotyping was routine, and standard management was to assign and raise all children with CAH according to their gonads and karyotypes, no matter how virilized. Markedly virilized girls were usually referred to a pediatric surgeon, often a pediatric urologist for a reconstructive vaginoplasty and clitoral reduction or recession—surgery to create or enlarge a vaginal opening and reduce the size or protrusion of the clitoris. This approach was designed to preserve fertility for both sexes and remains the standard management, but two aspects of this management have been challenged: assignment of completely virilized genetic females and the value and age of corrective surgery.[80][77]
The first questions about assignment were raised in the early 1980s when Money and others reported an unexpectedly high rate of failure to achieve normal adult sexual relationships (i.e., heterosexual orientation, marriage, and children) in grown women with CAH (though all had female gender identities). However, the sample was small, and results seemed interpretable in many ways: selection bias, early hormone effects on orientation, or sexual dysfunction created by residual body abnormalities or by the genital surgery itself. From a perspective two decades later, the report was one of the first pieces of evidence that the standard management paradigm was not always producing hoped-for outcomes.[80][81]
Despite these concerns, no significant opposition to standard management arose until the mid-1990s, when a confluence of evidence and opinion from several sources led to a re-examination of outcomes. Several intersex support and advocacy groups (e.g., the Intersex Society of North America) began to publicly criticize infant genital surgery based on unsatisfactory outcomes of some adults who had been operated on as infants. Their complaints were that they had reduced ability to enjoy sexual relations or that they resented not having had the choice of gender assignment or surgical reconstruction left until they were old enough to participate.[82][83]
In 1997, influential articles by Reiner, Diamond, and Sigmundson advocated consideration of (1) male sex assignment in the unambiguously male XX infants (most of whom are considered male until the CAH is recognized at 1–2 weeks of age), and (2) delaying reconstructive surgery until the patient is old enough to participate in the decision.[63]
Although the standard management approach remains "standard", more time and consideration are being given in many cases to explaining alternatives to parents and a small number of XX children with unambiguously male external genitalia are again being raised as boys.[84][85][86][87][88]
In the milder, nonclassical form of CAH, the androgen excess is subtle enough that virilization is not apparent or goes unrecognized at birth and in early childhood. However, androgen levels are above normal and slowly rise during childhood, producing noticeable effects between 2 and 9 years of age.[89]
Appearance of pubic hair in mid-childhood is the most common feature that leads to evaluation and diagnosis of the late onset (nonclassical) CAH. Other accompanying features are likely to be tall stature and accelerated bone age (often 3–5 years ahead). Often present are increased muscle mass, acne, and adult body odor. In boys the penis will be enlarged. Mild clitoral enlargement may occur in girls, and sometimes a degree of prenatal virilization is recognized that may have gone unnoticed in infancy.[89][90]
The principal goals of treatment of nonclassical CAH are to preserve as much growth as possible and to prevent central precocious puberty if it has not already been triggered. These are more difficult challenges than in CAH detected in infancy because moderate levels of androgens will have had several years to advance bone maturation and to trigger central puberty before the disease is detected.[91][89]
A diagnosis of nonclassical CAH is usually confirmed by discovering extreme elevations of 17OHP along with moderately high testosterone levels. A cosyntropin stimulation test may be needed in mild cases, but usually the random levels of 17OHP are high enough to confirm the diagnosis.[92][93][94]
Elevated 17OHP may activate androgen "backdoor" pathway, that leads to excess of 5α-dihydrotestosterone and other potent androgens.[95][96]
The mainstay of treatment of symptomos of hyperandrogenism is suppression of androgens of adrenal origin by a glucocorticoid such as hydrocortisone. Mineralocorticoid is only added in cases where the plasma renin activity is high.[97][98][99] Most patients that have the nonclassic form do not require any glucocorticoid replacement therapy,[4] as cortisol synthesis impairment is mild but clinically silent.[100][101][12] Patients usually have the same baseline but lower peak cortisol levels comparing to healthy controls.[102][103] The exact degree of cortisol synthesis impairment depends on genotype.[104]
An important aspect of management of nonclassic CAH is suppression of central precocious puberty if it has begun. The usual clues to central puberty in boys are that the testes are pubertal in size, or that androgens of adrenal origin remain elevated even when the 17OHP has been reduced toward normal. In girls central puberty is less often a problem, but breast development would be the main clue. Central precocious puberty is suppressed when appropriate by leuprolide.[105][106][107]
As outlined above, recent additions to treatment to preserve growth include aromatase inhibition to slow bone maturation by reducing the amount of testosterone converted to estradiol, and use of blockers of estrogen for the same purpose.[108][109][110][111]
Once adrenal suppression has been achieved, the patient needs stress steroid coverage as described above for significant illness or injury.[112]
Various variants of the CYP21A2 gene result various degrees of hyperandrogenism, some of which may be such a mild that they may not even cause problems in males and may not be recognized until adolescence or later in females.[113] Mild androgen effects in young women may include hirsutism, acne, or anovulation (which in turn can cause infertility). Testosterone levels in these women may be mildly elevated, or simply above average. Despite the prevailing notion that testosterone and 5α-dihydrotestosterone (DHT) are the primary human androgens, this notion only applies to healthy men.[114] Although testosterone has been traditionally used as a biomarker of hyperandrogenism,[115] it correlates poorly with clinical measurements of androgen excess.[27][114] While testosterone levels can appear normal, ignoring the alternative androgen pathways may lead to diagnostic errors since hyperandrogenism may be caused by potent androgens such as DHT produced by a backdoor pathway and 11-oxygenated androgens.[27][116][117] It may be that 11-ketotestosterone is the main androgen in women since it circulates at similar level to testosterone and may[118] or may not[119][120] decline with age as testosterone does. While 11-ketodihydrotestosterone (11KDHT) is equipotent to DHT, circulating levels of 11KDHT are lower than DHT.[27] Unlike testosterone and androstenedione, 11-oxygenated androgens are not known to be aromatized to estrogens in the human body.[27][42][121]
These clinical features are those of polycystic ovary syndrome (PCOS), and a small percentage of women with PCOS are found to have late onset CAH when investigated. Late onset CAH is often misdiagnosed as PCOS.[122]
Late onset CAH is often diagnosed in the context of infertility assessment in women. Diagnosis of late onset CAH may be suspected from a high 17OHP level, but some cases are so mild that the elevation is only demonstrable after cosyntropin stimulation. Treatment may involve a combination of very low dose glucocorticoid to reduce adrenal androgen production and any of various agents to block the androgen effects and/or induce ovulation. During the follicular phase of the menstrual cycle, progesterone accumulates along with 17OHP which can thin the endometrium and change cervical mucus in a manner similar to the effect of progestogen contraceptives, interferes with the normal menstrual cycle, which can lead to oligomenorrhea or amenorrhea[12] and impairs sperm penetration.[123] Abnormal endometrial development leads to decreased uterine receptivity, which also contributes to infertility.[124] Once attempting to conceive, most women with late onset CAH will become pregnant within a year with or without treatment, but they have an increased risk of miscarriage.[125][12]
Late onset CAH was originally characterized in 1957 by French biochemist Jacques Decourt,[126] but the association with mild 21-hydroxylase deficiency called nonclassical 21-hydroxylase deficiency, which is characterized by diverse hyperandrogenic symptoms appearing postnatally in males and females, was first described in 1979 by Maria New.[127] New since then have studied ways to reduce androgen excess and found out that treatment with dexamethasone 0.25mg orally every evening reversed acne and irregular menstruation in 3 months, but hirsutism required up to 30 months.[128][129] Dexamethasone has glucocorticoid activity, and potent ACTH-suppression properties within the hypothalamic–pituitary–adrenal axis.[130][131] Lower ACTH leads to reduced production of all the steroids, including androgens. According to 2018 Clinical Practice Guideline, glucocorticoid treatment is not recommended in asymptomatic individuals, however, if the symptoms of androgen excess are sufficient, dexamethasone treatment may be prescribed.[4] Another treatment option is oral contraceptive pills.[132][100]
The CYP21A2 gene for the P450c21 enzyme (also known as 21-hydroxylase) is at 6p21.3,[133] amid genes HLA B and HLA DR coding for the major human histocompatibility loci (HLA). The protein-coding CYP21A2 gene may be accompanied with one or several copies of the nonfunctional pseudogeneCYP21A1P.[134][36] Scores of abnormal alleles of CYP21A2 have been documented, most arising from recombinations of homologous regions of CYP21A2 and CYP21A1P.[36] The 21-hydroxylase deficiency may be caused by macrodeletions of about 30 Kb, which includes not only most of the 5′ region of the CYP21A2 gene, but also all of the C4B gene and 3′ regions of the CYP21A1P pseudogene. Duplications of CYP21A1P pseudogene and C4B gene are often associated with nonclassic 21-hydroxylase deficiency.[33]
Due to the high degree of homology between the CYP21A2 gene and the CYP21A1P pseudogene, and the complexity of the locus, research on the molecular level is difficult.[135]
Differences in residual enzyme activity of the various alleles account for the various degrees of severity of the disease.[136][137][138] Inheritance of all forms of 21-hydroxylase CAH is autosomal recessive,[4] except some mild disease-causing variants such as p.V281L that seem to exert dominant negative effects on enzymatic activity.[2]
Persons affected by any forms of the disease have two abnormal alleles, and both parents are usually heterozygotes (or carriers). When both parents carry an abnormal allele, each child has a 25% chance of having the disease, a 50% chance of being a carrier like the parents, and a 25% chance of having two normal genes.[4]
It is possible to test for heterozygosity by measuring 17OHP elevation after ACTH stimulation.[139]
More than 200 disease-causing variants within the CYP21A2 gene have been identified so far that lead to 21-hydroxylase deficiency.[140] Most patients have at least two of these variants present as compound heterozygous.[141][142][143]
There are some mild disease-causing variants that seem to exert dominant negative effects on enzymatic activity, even if present as single heterozygous. One example is a point mutation in exon 7 of CYP21A2, (p.V281L), which is commonly found in LOCAH-associated alleles.[2][135][142] Carriers for this mutation retain 20%–50% of 21-hydroxylase activity,[144][145] but are at higher risk of symptoms of androgen excess than carriers of the severe mutations,[146] and had higher adrenocorticotropic hormone (ACTH) stimulated 17OHP,[147] suggesting that the mutant protein V281L enzyme co-expressed with the wild-type (healthy) enzyme resulted in an apparent dominant negative effect on the enzymatic activity.[148]
There is a good correlation between the genotype and phenotype. As a result, the CYP21A2 genotyping has high diagnostic value. However, the genotyping of the CYP21A2 gene is prone to errors, especially due to the closely located and highly homologous pseudogene CYP21A1P and the complex duplications, deletions and rearrangements within the chromosome 6p21.3. That's why CYP21A2 genotyping, interpretation of the results, and adequate genetic counseling for patients and their families requires deep understanding of CYP21A2 genetics.[36]
The CYP21A1Ppseudogene retains 98% exonic sequence identity with the functional gene CYP21A2,[149][150] and the high degree of sequence similarity between them indicates that these two genes are evolving in tandem through intergenic exchange of DNA.[151] Both genes are located on chromosome 6, in the major histocompatibility complexIII (MHC class III)[152] close to the Complement component 4 genes C4A and C4B, the Tenascin X gene TNXB and STK19.[153] MHC class III is the most gene-dense region of the human genome, containing many genes that yet have unknown function or structure.[154][152]
The CYP21A2 gene is located within the RCCX cluster (an abbreviation composed of the names of the genes RP (a former name for STK19 serine/threonine kinase 19),[155][156]C4, CYP21 and TNX),[157] which is the most complex gene cluster in the human genome.[158] The number of RCCX segments varies between one and four in a chromosome,[155] with the prevalence of approximately 15% for monomodular, 75% for bimodular (STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB),[156][138] and 10% for trimodular in Europeans.[159] The quadrimodular structure of the RCCX unit is very rare.[160][155][159] In a monomodular structure, all of the genes are functional i.e. protein-coding, but if a module count is two or more, there is only one copy of each functional gene rest being non-coding pseudogenes with the exception of the C4 gene which always has active copies.[155][159]
Due to the high degree of homology between the CYP21A2 gene and the CYP21P1 pseudogene and the complexity of the locus, it is difficult to study the CYP21A2 gene at the molecular level, giving sometimes inaccurate or inconclusive results.[135] Targeted long-read sequencing method aims to improve the accuracy of the findings,[125] however, the clinical use of this method is limited.[161]
The cryptic form of (CAH) refers to a condition in which an individual is genetically determined to have the nonclassic variant of CAH but does not display any obvious symptoms. The term "cryptic" is used due to the lack of symptomatology in these individuals. Most males and some females with nonclassic CAH do not exhibit clinical signs or symptoms. However, the exact prevalence of asymptomatic individuals among those genetically identified with nonclassic CAH is currently unknown. Therefore, it is also uncertain what proportion of this population represents the cryptic form of CAH.[2]
Deficient activity of this enzyme reduces the efficiency of cortisol synthesis, with consequent hyperplasia of the adrenal cortex and elevation of ACTH levels. ACTH stimulates uptake of cholesterol and synthesis of pregnenolone. Steroid precursors up to and including progesterone, 17α-hydroxypregnenolone, and especially 17OHP accumulate in the adrenal cortex and in circulating blood. Blood levels of 17OHP can reach 10-1000 times the normal concentration.[163]
Since 21-hydroxylase activity is not involved in synthesis of androgens, a substantial fraction of the large amounts of 17α-hydroxypregnenolone is diverted to synthesis of DHEA, androstenedione, and other androgens of adrenal origin beginning in the third month of fetal life in both sexes.[164]
Synthesis of aldosterone is also dependent on 21-hydroxylase activity. Although fetal production is impaired, it causes no prenatal effects, as the placental connection allows maternal blood to "dialyze" the fetus and maintain both electrolyte balance and blood volume.[165]
Diagnosis
Since CAH is an autosomal recessive disease, most children with CAH are born to parents unaware of the risk and with no family history. Each child will have a 25% chance of being born with the disease.[4]
Classification
The condition can be classified into "salt-wasting", "simple virilizing", and "nonclassical" forms.[4]
Cortisol is mildly reduced depending on genotype,[104] but aldosterone is not.
Patients who are genetically found to have nonclassical CAH but are asymptomatic
No symptoms of androgen excess, levels of androgens are within the normal range.
Neither aldosterone nor cortisol are reduced.
The salt-wasting and simple virilizing types are sometimes grouped together as "classical".[166]
Newborn screening
Conditions justifying newborn screening for any disorder include (1) a simple test with an acceptable sensitivity and specificity, (2) a dire consequence if not diagnosed early, (3) an effective treatment if diagnosed, and (4) a frequency in the population high enough to justify the expense. In the last decade more states and countries are adopting newborn screening for salt-wasting CAH due to 21-hydroxylase deficiency, which leads to death in the first month of life if not recognized.[57][23]
The salt-wasting form of CAH has an incidence of 1 in 15,000 births and is potentially fatal within a month if untreated. Steroid replacement is a simple, effective treatment. However, the screening test itself is less than perfect, because of low specificity and high levels of false positives, meaning that the tests sometimes give incorrect results, saying the baby has CAH when they actually do not. This can make the family worried and spend money unnecessarily on hospital visits and extra tests that are not needed.[167] For the newborn screening, levels of 17α-hydroxyprogesterone (17OHP) are typically measured against predetermined cutoff, which depends on the measurement method.[163][57] While the 17OHP level is easy to measure and sensitive (rarely missing real cases), the test has a poorer specificity, giving high rates of false positives.[167] Screening programs in the United States have reported that 99% of positive screens turn out to be false positives upon investigation of the infant.[168][169][170] This is a higher rate of false positives than the screening tests for many other congenital metabolic diseases.[163][57]
Measurement 17OHP by LC-MS/MS reduces false positive rate in newborn screening in comparison to measurement by immunoassays. 17OHP steroid precursors and their sulphated conjugates which are present in the first two days after birth in healthy infants and longer in pre-term neonates, cross-react in immunoassays with 17OHP, giving falsely high 17OHP levels.[171][172]
When a positive result is detected, the infant must be referred to a pediatric endocrinologist to confirm or disprove the diagnosis. Since most infants with salt-wasting CAH become critically ill by 2 weeks of age, the evaluation must be done rapidly despite the high false positive rate.[173]
While 17OHP with or without ACTH stimulation is the main marker for 21-hydroxylase deficiency, other markers have been proposed, with various degrees of acceptance:[175][176]
21-Deoxycortisol is elevated in 21-hydroxylase deficiency.[177] However, it is not elevated in preterm infants or in other forms of congenital adrenal hyperplasia. Unlike 17OHP, 21-deoxycortisol is not produced in the gonads and is uniquely adrenal-derived.[12] Consequently, 21-deoxycortisol it is a more specific marker of the 21-hydroxylase deficiency than 17OHP. Even so, 21-deoxycortisol measurement has not been commonly performed by laboratories until 2019.[178][179] Still, 21-deoxycortisol can be a key screening marker for 21-hydroxylase deficiency in newborn screening.[177][180]
21-Deoxycorticosterone, also known as 11β-hydroxyprogesterone (11β-OHP), have been proposed as a marker in 1987.[181][182] A study in 2017 has shown that in subjects with 21-hydroxylase deficiency, serum 11β-OHP concentrations range from 0.012 to 3.37ng/mL, while in control group it was below detection limit of 0.012ng/mL.[38] This marker did not gain acceptance due to the fact that the test for the levels of this steroid is not routinely offered by diagnostic laboratories.[183]
Progesterone levels are higher in CAH subjects. A study has revealed that serum progesterone concentrations in boys (10 days to 18 years old) with 21-hydroxylase deficiency reached levels up to 10.14ng/mL, i.e. similar to female luteal values, while in the control group of boys average level was 0.07ng/mL (0.22 nmol/L), with values ranging from 0.05 to 0.40ng/mL.[38] The authors of the study propose to use progesterone as an additional marker for 21-hydroxylase deficiency. The study shows that the progesterone levels in CAH and non-CAH females are the same as in CAH and non-CAH males respectively – it is the condition that affects progesterone levels, not the sex, but for women between menarch and menopause, progesterone should be measured in days 3–5 of the cycle to have diagnostic value – the same condition also applies for 17OHP. The specificity of progesterone as a marker of 21-hydroxylase deficiency, as opposed to deficiency of other enzymes involved in steroid pathways, is still not well studied.[184][185][186]
Cortisol is one of the two main final products of 21-hydroxylase, and the deficiency of this enzyme may lead to a certain degree of cortisol deficiency. Cortisol levels are lower in CAH subjects, on average,[38] however, in milder cases cortisol levels can be normal, but, this has not been yet well studied. Cortisol measurement using immunoassays is prone to cross-reactivity with various substances including 21-deoxycortisol that raises due to 21-hydroxylase deficiency, leading to falsely high cortisol levels when the true cortisol is actually low.[187][188] The selectivity offered by liquid chromatography-tandem mass spectrometry (LC-MS/MS) has largely overcome these limitations.[189][190] As a result, the use of LC-MS/MS instead of immunoassays in cortisol measurement aims to provide greater specificity.[177][191]
11-Deoxycortisol is a direct product of 17α-hydroxyprogesterone, with 21-hydroxylase catalyzing the reaction, and an intermediate product towards cortisol pathway. Reduced 21-hydroxylase activity leads to decreased levels of 11-deoxycortisol, but not all laboratories specify minimum reference value it, since it is mostly used as a biomarker for 11β-hydroxylase deficiency, where 11-deoxycortisol levels increase dramatically, so the laboratories may only specify the maximum reference value.[175]
Treatment
Prenatal treatment
Clinical Practice Guideline advise that clinicians continue to regard prenatal therapy as experimental.[4]
Because the period during which fetal genitalia may become virilized begins about 6 weeks after conception, prenatal treatment to avoid virilization must be started by 6 to 7 weeks.[4]
Application of dexamethasone in prenatal treatment
Adrenal glands of female fetuses with CAH begin producing excess androgens[192] by the 9th week of gestation.[193][194] The most important aspects of virilization (urogenital closure and phallic urethra) occur between 8 and 12 weeks.[195] Theoretically, if enough glucocorticoid could be supplied to the fetus to reduce adrenal androgen biosynthesis by the 9th week, virilization could be prevented and the difficult decision about timing of surgery avoided.[193][196]
The challenge of preventing severe virilization of girls is twofold: detection of CAH at the beginning of the pregnancy, and delivery of an effective amount of glucocorticoid to the fetus without causing harm to the mother.[197][198][199]
The first problem has not yet been entirely solved, but it has been shown that if dexamethasone is taken by a pregnant woman, enough can cross the placenta to suppress fetal adrenal function.[200]
At present no program screens for risk in families who have not yet had a child with CAH. For families desiring to avoid virilization of a second child, the current strategy is to start dexamethasone as soon as a pregnancy has been confirmed even though at that point the chance that the pregnancy is a girl with CAH is only 12.5%. Dexamethasone is taken by the mother each day until it can be safely determined whether she is carrying an affected girl.[201]
Whether the fetus is an affected girl can be determined by chorionic villus sampling at 9–11 weeks of gestation, or by amniocentesis at 15–18 weeks gestation. In each case the fetal sex can be determined quickly, and if the fetus is a male the dexamethasone can be discontinued. If female, fetal DNA is analyzed to see if she carries one of the known abnormal alleles of the CYP21 gene. If so, dexamethasone is continued for the remainder of the pregnancy at a dose of about 1mg daily.[201]
Most mothers who have followed this treatment plan have experienced at least mild cushingoid effects from the glucocorticoid but have borne daughters whose genitalia are much less virilized.[201]
Dexamethasone is used as an off-label early prenatal treatment for the symptoms of CAH in female fetuses, but it does not treat the underlying congenital disorder. A 2007 Swedish clinical trial found that treatment may cause cognitive and behavioural defects, but the small number of test subjects means the study cannot be considered definitive. A 2012 American study found no negative short-term outcomes, but "lower cognitive processing in CAH girls and women with long-term dexamethasone exposure."[202] Administration of pre-natal dexamethasone has been the subject of controversy over issues of informed consent and because treatment must predate a clinical diagnosis of CAH in the female fetus,[203] especially because in utero dexamethasone may cause metabolic problems that are not evident until later in life; Swedish clinics ceased recruitment for research in 2010.[204]
The treatment has also raised concerns in LGBT and bioethics communities following publication of an essay posted to the forum of the Hastings Center, and research in the Journal of Bioethical Inquiry, which found that pre-natal treatment of female fetuses was suggested to prevent those fetuses from becoming lesbians after birth, may make them more likely to engage in "traditionally" female-identified behaviour and careers, and more interested in bearing and raising children. Citing a known attempt by a man using his knowledge of the fraternal birth order effect to avoid having a homosexual son by using a surrogate, the essayists (Professor Alice Dreger of Northwestern University's Feinberg School of Medicine, Professor Ellen Feder of American University and attorney Anne Tamar-Mattis) suggest that prenatal dexamethasone treatments constitute the first known attempt to use in utero protocols to reduce the incidence of homosexuality and bisexuality in humans.[205][206] Research on the use of prenatal hormone treatments to prevent homosexuality stretches back to the early 1990s or earlier.[207]
Since CAH is a recessive gene, both the mother and father must be recessive carriers of CAH for a child to have CAH. Due to advances in modern medicine, those couples with the recessive CAH genes have an option to prevent CAH in their offspring through preimplantation genetic diagnosis (PGD). In PGD, the egg is fertilized outside the woman's body in a Petri dish (IVF). On the 3rd day, when the embryo has developed from one cell to about 4 to 6 cells, one of those cells is removed from the embryo without harming the embryo. The embryo continues to grow until day 5 when it is either frozen or implanted into the mother. Meanwhile, the removed cell is analyzed to determine if the embryo has CAH. If the embryo is determined to have CAH, the parents may make a decision as to whether they wish to have it implanted in the mother or not.[192]
Meta-analysis of the studies supporting the use of dexamethasone on CAH at-risk fetuses found "less than one half of one percent of published 'studies' of this intervention were regarded as being of high enough quality to provide meaningful data for a meta-analysis. Even these four studies were of low quality... in ways so slipshod as to breach professional standards of medical ethics"[206] and "there were no data on long-term follow-up of physical and metabolic outcomes in children exposed to dexamethasone".[208]
Long-term management of CAH
Management of infants and children with CAH is complex and warrants long-term care in a pediatric endocrine clinic. After the diagnosis is confirmed, and any salt-wasting crisis averted or reversed, major management issues include:[citation needed]
Optimizing androgen suppression and fertility in women with CAH
Management of adults with CAH should take into consideration side effects of long-term glucocorticoid exposure, such as cardiovascular morbidities.[99]
Glucocorticoids are provided to all children and adults with all but the mildest and latest-onset forms of CAH. The glucocorticoids provide a reliable substitute for cortisol, thereby reducing ACTH levels. Reducing ACTH also reduces the stimulus for continued hyperplasia and overproduction of androgens. In other words, glucocorticoid replacement is the primary method of reducing the excessive adrenal androgen production in both sexes. A number of glucocorticoids are available for therapeutic use. Hydrocortisone or liquid prednisolone is preferred in infancy and childhood, and prednisone or dexamethasone are often more convenient for adults.[209][176]
The glucocorticoid dose is typically started at the low end of physiologic replacement (6–12mg/m2)[4] but is adjusted throughout childhood to prevent both growth suppression from too much glucocorticoid and androgen escape from too little. Serum levels of 17OHP, testosterone, androstenedione, and other adrenal steroids are followed for additional information, but may not be entirely normalized even with optimal treatment. As regular monitoring is needed, patients can be monitored non-invasively by measuring 17OHP and androstenedione in saliva.[211] (See Glucocorticoid for more on this topic.) However, the currently used glucocorticoid therapy methods may lead to unphysiological doses that, in addition to the problems caused by overexposure of androgens, can harm health. Various clinical results, besides the steroids, require regular monitoring. The negative consequences are primarily the result of non-physiological glucocorticoid replacement.[212] During illness or physical or mental stress, the demand for cortisol can increase, sometimes leading to adrenal crises. As current glucocorticoid therapy regiments fail to replicate the physiologic circadian rhythm and the glucocorticoids are often administered at a higher dose to treat androgen excess, the long-term consequences of the therapy, such as decreased bone density and an increased cardiometabolic risk profile,[99] should be addressed; other consequences of CAH such as impaired quality of life and affected mental health should be taken into consideration; periodic assessments and monitoring of patients should take place in specialized centers to detect and treat potential complications early.[136] Clinical studies are underway to find ways to better mimic the normal circadian rhythm of cortisol secretion and to reduce the total dose of glucocorticoids.[213]
Mineralocorticoids are replaced in all infants with salt-wasting and in most patients with elevated renin levels. Fludrocortisone is the only pharmaceutically available mineralocorticoid and is usually used in doses of 0.05 to 2mg daily.[4]Electrolytes, renin, and blood pressure levels are followed to optimize the dose.[214]
Even after diagnosis and initiation of treatment, a small percentage of children and adults with infancy or childhood onset CAH die of adrenal crisis.[4] Deaths from this are entirely avoidable if the child and family understand that the daily glucocorticoids cannot be allowed to be interrupted by an illness. When a person is well, missing a dose, or even several doses, may produce little in the way of immediate symptoms. However, glucocorticoid needs are increased during illness and stress, and missed doses during an illness such as the ifluenza or viral gastroenteritis can lead within hours to reduced blood pressure, shock, and death.[215][216]
To prevent this, all persons taking replacement glucocorticoids are taught to increase their doses in the event of illness, surgery, severe injury, or severe exhaustion. More importantly, they are taught that vomiting warrants an injection within hours of hydrocortisone (e.g., SoluCortef) or other glucocorticoid. This recommendation applies to both children and adults. Because young children are more susceptible to vomiting illnesses than adults, pediatric endocrinologists usually teach parents how to give hydrocortisone injections.[216][209]
As an additional precaution, persons with adrenal insufficiency are advised to wear a medical identification tag or carry a wallet card to alert those who may be providing emergency medical care of the urgent need for glucocorticoids.[217]
Reconstructive surgery
Surgery need never be considered for genetically male (XY) infants because the excess androgens do not produce anatomic abnormality. However, surgery for severely virilized XX infants is often performed and has become a subject of debate.[218]
Surgical reconstruction of abnormal genitalia has been offered to parents of severely virilized girls with CAH since the first half of the 20th century. The purposes of surgery have generally been a combination of the following:[219]
To make the external genitalia look more female than male;
To make it possible for these girls to participate in normal sexual intercourse when they grow up;
To improve their chances of fertility;
To reduce the frequency of urinary infections.[219]
In the 1950s and 1960s, surgery often involved clitorectomy (removal of most of the clitoris), an operation that also reduced genital sensation. In the 1970s, new operative methods were developed to preserve innervation and clitoral function. However, a number of retrospective surveys in the last decade suggest that (1) sexual enjoyment is reduced in many women even after nerve-sparing procedures, and (2) women with CAH who have not had surgery also have a substantial rate of sexual dysfunction. (See Intersex surgery for an overview of procedures and potential complications, and History of intersex surgery for a fuller discussion of the controversies.) Many patient advocates and surgeons argue for deferring surgery until adolescence or later, while some surgeons continue to argue that infant surgery has advantages.[220]
Optimizing growth in CAH
It is unclear what concentration of adrenal androgens is best for normal growth, puberty, and bone health.[221]
One of the challenging aspects of long-term management is optimizing growth so that a child with CAH achieves his or her height potential because both undertreatment and overtreatment can reduce growth or the remaining time for growth. While glucocorticoids are essential for health, dosing is always a matter of approximation. In even mildly excessive amounts, glucocorticoids slow growth. On the other hand, adrenal androgens are readily converted to estradiol, which accelerates bone maturation and can lead to early epiphyseal closure. This narrow target of optimal dose is made more difficult to obtain by the imperfect replication of normal diurnal plasma cortisol levels produced by 2 or 3 oral doses of hydrocortisone.[222][223] As a consequence, average height losses of about 4inches (10cm) have been reported with traditional management.[4]
Traditionally, pediatric endocrinologists have tried to optimize growth by measuring a child every few months to assess current rate of growth, by checking the bone age every year or two, by periodically measuring 17OHP and testosterone levels as indicators of adrenal suppression, and by using hydrocortisone for glucocorticoid replacement rather than longer-acting prednisone or dexamethasone.[163]
The growth problem is even worse in the simple virilizing forms of CAH which are detected when premature pubic hair appears in childhood, because the bone age is often several years advanced at the age of diagnosis. While a boy (or girl) with simple virilizing CAH is taller than peers at that point, he will have far fewer years remaining to grow, and may go from being a very tall 7-year-old to a 62-inch 13-year-old who has completed growth. Even with adrenal suppression, many of these children will have already had central precocious puberty triggered by the prolonged exposure of the hypothalamus to the adrenal androgens and estrogens. If this has begun, it may be advantageous to suppress puberty with a gonadotropin-releasing hormone agonist such as leuprolide to slow continuing bone maturation.[224][225][226]
In recent years some newer approaches to optimizing growth have been researched and are beginning to be used. It is possible to reduce the effects of androgens on the body by blocking the receptors with an antiandrogen such as flutamide and by reducing the conversion of testosterone to estradiol. This conversion is mediated by aromatase and can be inhibited by aromatase blockers such as testolactone. Blocking the effects and conversions of estrogens will allow use of lower doses of glucocorticoids with less risk of acceleration of bone maturation. Other proposed interventions have included bilateral adrenalectomy to remove the androgen sources, or growth hormone treatment to enhance growth.[227]
Preventing hyperandrogenism and optimizing fertility
As growth ends, management in girls with CAH changes focus to optimizing reproductive function. Both excessive androgens from the adrenals and excessive glucocorticoid treatment can disrupt ovulation, resulting in irregularity of menses or amenorrhea, as well as infertility. Continued monitoring of hormone balance and careful readjustment of glucocorticoid dose can usually restore fertility, but as a group, women with CAH have a lower fertility rate than a comparable population.[4]
Classical CAH has little effect on male fertility unless an adult stops taking his glucocorticoid medication entirely for an extended time, in which case excessive adrenal androgens may reduce testicular production as well as spermatogenesis.[4] Regarding nonclassic CAH, glucocorticoid therapy is typically not necessary for lifelong management. However, in certain cases, this treatment may be required to address fertility issues such as oligomenorrhea, amenorrhea, or difficulty conceiving. About 90% of women with nonclassic CAH remain undiagnosed. Once attempting to conceive, approximately 83% of women win nonclassic CAH successfully achieve pregnancy within one year, irrespective of whether they receive glucocorticoid therapy or not, but the therapy reduces risk of miscarriage.[12]
Psychosexual development and issues
Nearly all mammals display sex-dimorphic reproductive and sexual behavior (e.g., lordosis and mounting in rodents). Much research has made it clear that prenatal and early postnatal androgens play a role in the differentiation of most mammalian brains. Experimental manipulation of androgen levels in utero or shortly after birth can alter adult reproductive behavior.[4]
Girls and women with CAH constitute the majority of genetic females with normal internal reproductive hormones who have been exposed to male levels of androgens throughout their prenatal lives. Milder degrees of continuing androgen exposure continue throughout childhood and adolescence as a consequence of the imperfections of current glucocorticoid treatment for CAH. The psychosexual development of these girls and women has been analyzed as evidence of the role of androgens in human sex-dimorphic behaviors.[4]
In comparison to unaffected individuals, patients with classical CAH exhibit a greater prevalence of anxiety disorders, depressive disorders, alcohol misuse, personality disorders, and suicidal tendencies in males, while females are more prone to adjustment disorders.[12] In comparison to the general population, individuals with CAH have a higher prevalence of substance abuse and attention deficit–hyperactivity disorder (ADHD). This is particularly observed in females with the most severe null genotype and males who receive a diagnosis after the neonatal period.[12]
Girls with CAH have repeatedly been reported to spend more time with "sex-atypical" toys and "rough-and-tumble" play than unaffected sisters. These differences continue into adolescence, as expressed in social behaviors, leisure activities, and career interests. Interest in babies and becoming mothers is significantly lower by most measures.[12] Avoidance of romantic relationships is common in women and men with classical CAH.[12]
Gender identity of girls and women with CAH is most frequently observed to be female. Sexual orientation is more mixed, though the majority are heterosexual.[12] In one study, 27% of women with CAH were rated as bisexual in their orientations.[228]
A 2020 survey of 57 females with life-long experience of CAH and 132 parents of females with CAH in the United States revealed that majority of participants do not consider females with CAH to be intersex, and oppose a legal intersex designation of females with CAH.[229]
Cognitive effects are less clear. Altered fetal and postnatal exposure to androgens, as well as glucocorticoid therapy, affect brain development and function. Compared to healthy girls, those with classical CAH have more aggressive behavior but have better spatial navigation abilities, and the amygdala activation patterns differ between affected and healthy girls. Glucocorticoid therapy in CAH impairs working memory and causes brain changes, including white matter hyperintensities, suggesting a reduction in white matter structural integrity.[12] Cognitive impairment, when observed, has occasionally been attributed to hypoglycemia and electrolyte imbalances at the initial presentation.[12] A 2023 population-based cohort study of 714 patients with classical CAH and 71400 control subjects predict that patients with CAH have an increased prevalence of injuries and accidents, especially females, but the cause is not known.[230]
The increased prevalence of depressive and anxiety disorders, along with the prescription of antidepressants by medical practitioners, among youth with CAH in comparison to the general population indicates the potential necessity for screening for symptoms of depression and anxiety in this specific population.[231]
Given that people with CAH exhibit a higher incidence and severity of behavioral symptoms,[12] and that the manifestation of these behavioral problems can be attributed to excessive endogenous androgen exposure and side effects of glucucorticoid therapy,[12] emphasis should be placed on early optimization of CAH management to mitigate the psychological ramifications that may ensue.[232]
Prognosis
The outcomes and prognosis for CAH (Congenital Adrenal Hyperplasia) can vary depending on several factors, such as the specific type of CAH, its severity, early detection, and proper management. With appropriate medical care and ongoing treatment, individuals with CAH can lead healthy lives. Consistent monitoring and adherence to treatment are crucial for optimal outcomes.[4]
Children with CAH often experience increased height during early childhood, but their final adult height tends to be shorter than expected. Advanced bone age and early fusion of growth plates due to excess androgens contribute to this outcome. Additionally, glucocorticoid treatment for CAH can affect growth and result in decreased final height. Experimental treatments using growth hormone and luteinizing hormone-releasing hormone analogs have shown some success in increasing average height gains by approximately 7.3cm. Most children with CAH have normal neuropsychological development, although there may be differences in gendered play activities influenced by prenatal sex steroid exposure.[1]
While bone density is typically normal in most patients, metabolic abnormalities such as obesity, insulin resistance, dyslipidemia (abnormal blood lipid levels), and polycystic ovarian syndrome are more prevalent either due to the disease itself or long-term glucocorticoid treatment.[4]
Fertility is possible for both males and females with CAH but may be reduced due to multiple factors encompassing biology, psychology, social influences, and sexual aspects.[4]
Management of CAH requires collaboration among different subspecialty professionals considering the complex nature of the disease's complications and long-term consequences.[1]
Incidence and variants
According to most studies, the global incidence of classical forms range from about 1:14,000 to 1:18,000 births, based on newborn screening programs and national case registries, but this situation is more common in small genetically isolated populations with small gene pools.[4] 10% of patients with classical forms of CAH have CAH-X syndrome, that is marked by the presence of CAH features along with features indicative of hypermobility-type Ehlers-Danlos syndrome. This condition results from a contiguous gene deletion that impacts both CYP21A2 and TNXB genes.[233][234]
The incidence of nonclassical forms of CAH is 1:200 to 1:1000 based on various estimates, and is also higher in groups people with a high rate of marriage between relatives, up to 1:50.[4][235][128] Nonclassic CAH may be identified incidentally during the assessment of oligomenorrhea, amenorrhea, and infertility in females. However, a significant proportion, approximately 90%, of women with nonclassic CAH remain undiagnosed. As for men with nonclassic CAH, their asymptomatic nature commonly leads to underdiagnosis.[12]
History
In 19th Century, cases of CAH were reported, leading to the understanding that the adrenal gland influenced sexual phenotypes as well as being mysteriously required for survival.[236]
In 1865, CAH in its non-salt-wasting form was thoroughly described for the first time by a Neapolitan pathologist Luigi De Crecchio.[237] However, significant strides in understanding and treating the condition have been made over the years.[citation needed]
In the early 20th Century, numerous adrenal steroids were isolated, and bioassays eventually distinguished glucocorticoids, mineralocorticoids, and androgens.[236]
In 1950, Wilkins, Bartter, and Albright revolutionized clinical endocrinology with the use of cortisone to treat CAH and ushered in a productive era of pediatric adrenal research. Through careful clinical research, Wilkins established modern methods of treating CAH.[236]
In 1957, Alfred Bongiovanni noticed a defect in 21-hydroxylation in CAH, followed by defects in 3β-hydroxysteroid dehydrogenase and 11β-hydroxylase.[236]
P450 enzymes were described from 1962 to 1964, and 21-hydroxylation was the first identified activity of P450s.[236]
In the last quarter of the 20th Century, notable scholars who have contributed to the development of CAH knowledge include Hans Selye, who studied stress responses and adrenal function,[236] as well as Maria New, who conducted extensive research on prenatal treatment options for CAH.
From 1984 to 2004, the techniques of molecular genetics were applied to elucidate the genetic and biochemical bases of CAH.[236]
Society and culture
Society's understanding of CAH can vary widely, leading to different perceptions and attitudes toward individuals with the condition. Lack of awareness or misconceptions about CAH may contribute to stigma or discrimination.[238][239]
Discussions around intersex conditions have evolved over time as societal understanding has progressed. There is consensus that classical CAH is an intersex condition. However, for the nonclassical CAH, there is a debate on whether it can be considered an intersex condition. Anne Fausto-Sterling, an American sexologist, in a 2000 book "Sexing the Body" came up with an estimate that people with intersex conditions account for 1.7% of the general population.[240] This estimate is cited by a number of prominent intersex advocacy organizations.[241][242][243][244] Of these intersex individuals, according to Fausto-Sterling, 88% have nonclassical CAH.[240]Leonard Sax, an American psychologist and a family physician, criticized these figures in a review published in 2002 in The Journal of Sex Research, stating that from the clinician's perspective, nonclassical CAH is not an intersex condition.[245] Including nonclassical CAH in intersex prevalence estimates has been cited by Joel Best, is a professor of sociology and criminal justice, in a 2013 book "Stat-Spotting: A Field Guide to Identifying Dubious Data" as an example of misleading statistical practice.[246]
Different cultural contexts may shape views on gender identity, sexuality, and medical interventions for intersex conditions. Individuals with visible differences or unique health needs associated with CAH may face social stigmatization due to a lack of awareness, prejudice, or preconceived notions about intersexuality.[247][248][249][250]
Gene therapy is an experimental therapeutic approach that aims to correct the underlying genetic defect responsible for a particular condition. In the context of CAH, caused by mutations in the CYP21A2 gene, gene replacement therapy involves introducing a functional copy of this gene into the cells where this gene is expressed. The goal is to normalize the production of 21-hydroxylase, the enzyme encoded by CYP21A2. Providing a working copy of this gene may improve adrenal hormone synthesis and subsequently normalize cortisol and aldosterone production.[253]
Currently, gene replacement therapy for CAH is still at an early stage of research and development. Various approaches are being explored, including using viral vectors carrying a healthy copy of the CYP21A2 gene to deliver it into target cells. Another approach is based on utilizing human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs) to generate steroid-producing cells that can potentially replace or supplement dysfunctional adrenal tissue in individuals with CAH. However, it remains unclear how new approaches can be tested, as there are even no experimental models yet for this disease for gene therapy.[254]
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by impaired cortisol synthesis. It results from the deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex. Most of these disorders involve excessive or deficient production of hormones such as glucocorticoids, mineralocorticoids, or sex steroids, and can alter development of primary or secondary sex characteristics in some affected infants, children, or adults. It is one of the most common autosomal recessive disorders in humans.
Adrenal insufficiency is a condition in which the adrenal glands do not produce adequate amounts of steroid hormones. The adrenal glands—also referred to as the adrenal cortex—normally secrete glucocorticoids, mineralocorticoids, and androgens. These hormones are important in regulating blood pressure, electrolytes, and metabolism as a whole. Deficiency of these hormones leads to symptoms ranging from abdominal pain, vomiting, muscle weakness and fatigue, low blood pressure, depression, mood and personality changes to organ failure and shock. Adrenal crisis may occur if a person having adrenal insufficiency experiences stresses, such as an accident, injury, surgery, or severe infection; this is a life-threatening medical condition resulting from severe deficiency of cortisol in the body. Death may quickly follow.
Lipoid congenital adrenal hyperplasia is an endocrine disorder that is an uncommon and potentially lethal form of congenital adrenal hyperplasia (CAH). It arises from defects in the earliest stages of steroid hormone synthesis: the transport of cholesterol into the mitochondria and the conversion of cholesterol to pregnenolone—the first step in the synthesis of all steroid hormones. Lipoid CAH causes mineralocorticoid deficiency in affected infants and children. Male infants are severely undervirilized causing their external genitalia to look feminine. The adrenals are large and filled with lipid globules derived from cholesterol.
Congenital adrenal hyperplasia due to 11β-hydroxylase deficiency is a form of congenital adrenal hyperplasia (CAH) which produces a higher than normal amount of androgen, resulting from a defect in the gene encoding the enzyme steroid 11β-hydroxylase (11β-OH) which mediates the final step of cortisol synthesis in the adrenal. 11β-OH CAH results in hypertension due to excessive mineralocorticoid effects. It also causes excessive androgen production both before and after birth and can virilize a genetically female fetus or a child of either sex.
Congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene for one of the key enzymes in cortisol synthesis by the adrenal gland, 3β-hydroxysteroid dehydrogenase (3β-HSD) type II (HSD3B2). As a result, higher levels of 17α-hydroxypregnenolone appear in the blood with adrenocorticotropic hormone (ACTH) challenge, which stimulates adrenal corticosteroid synthesis.
Congenital adrenal hyperplasia due to 17α-hydroxylase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene CYP17A1, which produces the enzyme 17α-hydroxylase. It causes decreased synthesis of cortisol and sex hormones, with resulting increase in mineralocorticoid production. Thus, common symptoms include mild cortisol deficiency, ambiguous genitalia in men or amenorrhea at puberty in women, and hypokalemic hypertension. However, partial (incomplete) deficiency often has inconsistent symptoms between patients, and affected women may be asymptomatic except for infertility.
Hyperandrogenism is a medical condition characterized by high levels of androgens. It is more common in women than men. Symptoms of hyperandrogenism may include acne, seborrhea, hair loss on the scalp, increased body or facial hair, and infrequent or absent menstruation. Complications may include high blood cholesterol and diabetes. It occurs in approximately 5% of women of reproductive age.
17α-Hydroxyprogesterone (17α-OHP), also known as 17-OH progesterone (17-OHP), or hydroxyprogesterone (OHP), is an endogenous progestogen steroid hormone related to progesterone. It is also a chemical intermediate in the biosynthesis of many other endogenous steroids, including androgens, estrogens, glucocorticoids, and mineralocorticoids, as well as neurosteroids.
Steroid 21-hydroxylase is a protein that in humans is encoded by the CYP21A2 gene. The protein is an enzyme that hydroxylates steroids at the C21 position on the molecule. Naming conventions for enzymes are based on the substrate acted upon and the chemical process performed. Biochemically, this enzyme is involved in the biosynthesis of the adrenal gland hormones aldosterone and cortisol, which are important in blood pressure regulation, sodium homeostasis and blood sugar control. The enzyme converts progesterone and 17α-hydroxyprogesterone into 11-deoxycorticosterone and 11-deoxycortisol, respectively, within metabolic pathways which in humans ultimately lead to aldosterone and cortisol creation—deficiency in the enzyme may cause congenital adrenal hyperplasia.
Steroid 11β-hydroxylase, also known as steroid 11β-monooxygenase, is a steroid hydroxylase found in the zona glomerulosa and zona fasciculata of the adrenal cortex. Named officially the cytochrome P450 11B1, mitochondrial, it is a protein that in humans is encoded by the CYP11B1 gene. The enzyme is involved in the biosynthesis of adrenal corticosteroids by catalyzing the addition of hydroxyl groups during oxidation reactions.
An inborn error of steroid metabolism is an inborn error of metabolism due to defects in steroid metabolism.
Maria Iandolo New is a professor of Pediatrics, Genomics and Genetics at Icahn School of Medicine at Mount Sinai in New York City. She is an expert in congenital adrenal hyperplasia (CAH), a genetic condition affecting the adrenal gland that can affect sexual development.
21-Deoxycortisol, also known as 11β,17α-dihydroxyprogesterone or as 11β,17α-dihydroxypregn-4-ene-3,20-dione, is a naturally occurring, endogenous steroid related to cortisol (11β,17α,21-trihydroxyprogesterone) which is formed as a metabolite from 17α-hydroxyprogesterone via 11β-hydroxylase.
11β-Hydroxyprogesterone (11β-OHP), also known as 21-deoxycorticosterone, as well as 11β-hydroxypregn-4-ene-3,20-dione, is a naturally occurring, endogenous steroid and derivative of progesterone. It is a potent mineralocorticoid. Syntheses of 11β-OHP from progesterone is catalyzed by the steroid 11β-hydroxylase (CYP11B1) enzyme, and, to a lesser extent, by the aldosterone synthase enzyme (CYP11B2).
Adrenal steroids are steroids that are derived from the adrenal glands. They include corticosteroids, which consist of glucocorticoids like cortisol and mineralocorticoids like aldosterone, adrenal androgens like dehydroepiandrosterone (DHEA), DHEA sulfate (DHEA-S), and androstenedione (A4), and neurosteroids like DHEA and DHEA-S, as well as pregnenolone and pregnenolone sulfate (P5-S). Adrenal steroids are specifically produced in the adrenal cortex.
Walter L. Miller is an American endocrinologist and professor emeritus of pediatrics at the University of California, San Francisco (UCSF). Miller is expert in the field of human steroid biosynthesis and disorders of steroid metabolism. Over the past 40 years Miller's group at UCSF has described molecular basis of several metabolic disorders including, congenital adrenal hyperplasia, pseudo vitamin D dependent rickets, severe, recessive form of Ehlers-Danlos syndrome, 17,20 lyase deficiency caused by CYP17A1 defects, P450scc deficiency caused by CYP11A1 defects, P450 oxidoreductase deficiency.
Cytochrome P450 oxidoreductase deficiency (PORD) is a rare disease and inborn error of metabolism caused by deficiency of cytochrome P450 oxidoreductase (POR). POR is a 2-flavin protein that is responsible for the transfer of electrons from NADPH to all 50 microsomal cytochrome P450 (CYP450) enzymes. This includes the steroidogenic enzymes CYP17A1 (17α-hydroxylase/17,20-lyase), CYP19A1 (aromatase), and CYP21A2 (21-hydroxylase); CYP26B1 ; and the hepatic drug-metabolizing CYP450 enzymes, among many other CYP450 enzymes. Virilization of female infants in PORD may also be caused by alternative biosynthesis of 5α-dihydrotestosterone via the so-called "androgen backdoor pathway". The ABS component of severe forms of PORD is probably caused by CYP26B1 deficiency, which results in retinoic acid excess and defects during skeletal embryogenesis. All forms of PORD in humans are likely partial, as POR knockout in mice results in death during prenatal development.
Late onset congenital adrenal hyperplasia (LOCAH), also known as nonclassic congenital adrenal hyperplasia, is a milder form of congenital adrenal hyperplasia (CAH), a group of autosomal recessive disorders characterized by impaired cortisol synthesis that leads to variable degrees of postnatal androgen excess.
The androgen backdoor pathway is responsible for the synthesis of physiologically relevant androgens. This process starts with 21-carbon steroids, also known as pregnanes, and involves a step called "5α-reduction". Notably, this pathway does not require the intermediate formation of testosterone, hence the term "bypassing testosterone" is sometimes used in medical literature as the hallmark feature of this way of androgen biosynthesis. This feature is a key distinction from the conventional, canonical androgenic pathway, which necessitates the involvement of testosterone as an intermediate in the synthesis of androgens.
Crinecerfont is a corticotropin-releasing factor type 1 receptor (CRF1R) antagonist developed to treat classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency (21OHD). It is hoped to reduce the need for long-term, high dose glucocorticoid therapy and its associated adverse effects in people with CAH.
References
This article incorporates text available under the CC BY-SA 4.0 license.
↑ Dumić M, Brkljacić L, Mardesić D, Plavsić V, Lukenda M, Kastelan A (July 1985). "'Cryptic' form of congenital adrenal hyperplasia due to 21-hydroxylase deficiency in the Yugoslav population". Acta Endocrinol (Copenh). 109 (3): 386–92. doi:10.1530/acta.0.1090386. PMID2992207.
↑ Neves Cruz J, da Costa KS, de Carvalho TA, de Alencar NA (March 2020). "Measuring the structural impact of mutations on cytochrome P450 21A2, the major steroid 21-hydroxylase related to congenital adrenal hyperplasia". Journal of Biomolecular Structure & Dynamics. 38 (5): 1425–1434. doi:10.1080/07391102.2019.1607560. PMID30982438. S2CID115195169.
↑ Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland HJ, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Purnell J, Singer F, Stratakis CA, Trence DL, Wilson DP, New M, Yau M, Lekarev O, Lin-Su K, Parsa A, Pina C, Yuen T, Khattab A (15 March 2017). Congenital Adrenal Hyperplasia. MDText.com, Inc. PMID25905188.
↑ Varness TS, Allen DB, Hoffman GL (October 2005). "Newborn screening for congenital adrenal hyperplasia has reduced sensitivity in girls". The Journal of Pediatrics. 147 (4): 493–498. doi:10.1016/j.jpeds.2005.04.035. PMID16227036.
↑ Chan CL, McFann K, Taylor L, Wright D, Zeitler PS, Barker JM (July 2013). "Congenital adrenal hyperplasia and the second newborn screen". The Journal of Pediatrics. 163 (1): 109–13.e1. doi:10.1016/j.jpeds.2013.01.002. PMID23414665.
↑ Hindmarsh PC (April 2009). "Management of the child with congenital adrenal hyperplasia". Best Practice & Research. Clinical Endocrinology & Metabolism. 23 (2): 193–208. doi:10.1016/j.beem.2008.10.010. PMID19500763.
↑ Pang S, Clark A (1990). "Newborn screening, prenatal diagnosis, and prenatal treatment of congenital adrenal hyperplasia due to 21-hydroxylase deficiency". Trends in Endocrinology and Metabolism. 1 (6): 300–307. doi:10.1016/1043-2760(90)90068-e. PMID18411135. S2CID21880131.
↑ Barnard L, Gent R, van Rooyen D, Swart AC (November 2017). "Adrenal C11-oxy C21 steroids contribute to the C11-oxy C19 steroid pool via the backdoor pathway in the biosynthesis and metabolism of 21-deoxycortisol and 21-deoxycortisone". The Journal of Steroid Biochemistry and Molecular Biology. 174: 86–95. doi:10.1016/j.jsbmb.2017.07.034. PMID28774496. S2CID24071400.
↑ Kumar V, Abbas AK, Aster JC, Cotran RS, Robbins SL (2014). Robbins and Cotran pathologic basis of disease (Ninthed.). Philadelphia, PA: Elsevier/Saunders. p.1128. ISBN978-1-4557-2613-4. OCLC879416939.
↑ Murphy BE, Sebenick M, Patchell ME (January 1980). "Cortisol production and metabolism in the human fetus and its reflection in the maternal urine". J Steroid Biochem. 12: 37–45. doi:10.1016/0022-4731(80)90248-4. PMID7191459.
↑ Neunzig J, Milhim M, Schiffer L, Khatri Y, Zapp J, Sánchez-Guijo A, etal. (March 2017). "The steroid metabolite 16(β)-OH-androstenedione generated by CYP21A2 serves as a substrate for CYP19A1". The Journal of Steroid Biochemistry and Molecular Biology. 167: 182–191. doi:10.1016/j.jsbmb.2017.01.002. PMID28065637. S2CID36860068.
↑ van Rooyen D, Gent R, Barnard L, Swart AC (April 2018). "The in vitro metabolism of 11β-hydroxyprogesterone and 11-ketoprogesterone to 11-ketodihydrotestosterone in the backdoor pathway". The Journal of Steroid Biochemistry and Molecular Biology. 178: 203–212. doi:10.1016/j.jsbmb.2017.12.014. PMID29277707. S2CID3700135.
↑ Gueux B, Fiet J, Galons H, Boneté R, Villette JM, Vexiau P, etal. (January 1987). "The measurement of 11 beta-hydroxy-4-pregnene-3,20-dione (21-deoxycorticosterone) by radioimmunoassay in human plasma". primary. Journal of Steroid Biochemistry. 26 (1): 145–150. doi:10.1016/0022-4731(87)90043-4. PMID3546944.
1 2 Nagasaki K, Takase K, Numakura C, Homma K, Hasegawa T, Fukami M (November 2020). "Foetal virilisation caused by overproduction of non-aromatisable 11-oxygenated C19 steroids in maternal adrenal tumour". Human Reproduction. 35 (11): 2609–2612. doi:10.1093/humrep/deaa221. PMID32862221.
1 2 Rauh M, Gröschl M, Rascher W, Dörr HG (June 2006). "Automated, fast and sensitive quantification of 17 alpha-hydroxy-progesterone, androstenedione and testosterone by tandem mass spectrometry with on-line extraction". Steroids. 71 (6): 450–458. doi:10.1016/j.steroids.2006.01.015. PMID16569417. S2CID11605597.
↑ de Jesus LE, Costa EC, Dekermacher S (November 2019). "Gender dysphoria and XX congenital adrenal hyperplasia: how frequent is it? Is male-sex rearing a good idea?". Journal of Pediatric Surgery. 54 (11): 2421–2427. doi:10.1016/j.jpedsurg.2019.01.062. PMID30905417. S2CID85501467.
↑ Zwayne N, Chawla R, van Leeuwen K (August 2023). "Caring for Patients With Congenital Adrenal Hyperplasia Throughout the Lifespan". Obstetrics and Gynecology. 142 (2): 257–268. doi:10.1097/AOG.0000000000005263. PMID37473408. S2CID259996994.
↑ Amais DS, da Silva TE, Barros BA, de Andrade JG, de Lemos-Marini SH, de Mello MP, etal. (December 2022). "Sex dimorphism of weight and length at birth: evidence based on disorders of sex development". Annals of Human Biology. 49 (7–8): 274–279. doi:10.1080/03014460.2022.2134452. PMID36218438. S2CID252816435.
↑ Khattab A, Yau M, Qamar A, Gangishetti P, Barhen A, Al-Malki S, etal. (January 2017). "Long term outcomes in 46, XX adult patients with congenital adrenal hyperplasia reared as males". The Journal of Steroid Biochemistry and Molecular Biology. 165 (Pt A): 12–17. doi:10.1016/j.jsbmb.2016.03.033. PMID27125449. S2CID21521508.
1 2 Dessens AB, Slijper FM, Drop SL (August 2005). "Gender dysphoria and gender change in chromosomal females with congenital adrenal hyperplasia". Archives of Sexual Behavior. 34 (4): 389–397. doi:10.1007/s10508-005-4338-5. PMID16010462. S2CID11328239.
↑ Szymanski KM, Whittam B, Kaefer M, Frady H, Casey JT, Tran VT, etal. (April 2018). "Parental decisional regret and views about optimal timing of female genital restoration surgery in congenital adrenal hyperplasia". Journal of Pediatric Urology. 14 (2): 156.e1–156.e7. doi:10.1016/j.jpurol.2017.11.012. PMID29330019.
↑ Liao L, Sun M, Wang F, Pan S, Wu X, Huang X (December 2022). "Clinical characteristics of female patients diagnosed with congenital adrenal hyperplasia and plastic-esthetic related thoughts: a retrospective study". Neuro Endocrinology Letters. 43 (7–8): 378–384. PMID36720126.
↑ Zhang Q, Zang L, Zhang CY, Gu WJ, Li B, Jia XF, etal. (January 2022). "[Diagnosis and treatment of 21-hydroxylase deficiency with testicular adrenal rest tumors:a report of three cases and literature review]". Zhonghua Nei Ke Za Zhi (in Chinese). 61 (1): 72–76. doi:10.3760/cma.j.cn112138-20210718-00488 (inactive 2024-04-26). PMID34979773.{{cite journal}}: CS1 maint: DOI inactive as of April 2024 (link)
1 2 3 Orozco-Poore C, Keuroghlian AS (June 2023). "Neurological Considerations for "Nerve-Sparing" Cosmetic Genital Surgeries Performed on Children with XX Chromosomes Diagnosed with 21-Hydroxylase Congenital Adrenal Hyperplasia and Clitoromegaly". LGBT Health. 10 (8): 567–575. doi:10.1089/lgbt.2022.0160. PMID37319358. S2CID259173363.
↑ Money J (2000). "Outcome Research in Pediatric Psychoendocrinology and Sexology". Therapeutic Outcome of Endocrine Disorders. Springer New York, NY. pp.1–6. doi:10.1007/978-1-4612-1230-0_1. ISBN978-1-4612-7052-2.
↑ Bangalore Krishna K, Houk CP, Lee PA (June 2017). "Pragmatic approach to intersex, including genital ambiguity, in the newborn". Seminars in Perinatology. 41 (4): 244–251. doi:10.1053/j.semperi.2017.03.013. PMID28535943.
↑ Thyen U, Lanz K, Holterhus PM, Hiort O (2006). "Epidemiology and initial management of ambiguous genitalia at birth in Germany". Hormone Research. 66 (4): 195–203. doi:10.1159/000094782. PMID16877870. S2CID24768524.
↑ Ahmed SF, Rodie M (April 2010). "Investigation and initial management of ambiguous genitalia". Best Practice & Research. Clinical Endocrinology & Metabolism. 24 (2): 197–218. doi:10.1016/j.beem.2009.12.001. PMID20541148. S2CID205973113.
1 2 3 Brener A, Segev-Becker A, Weintrob N, Stein R, Interator H, Schachter-Davidov A, etal. (August 2019). "Health-Related Quality of Life in Children and Adolescents with Nonclassic Congenital Adrenal Hyperplasia". Endocrine Practice. 25 (8): 794–799. doi:10.4158/EP-2018-0617. PMID31013157. S2CID129942669.
↑ Labarta JI, Bello E, Ruiz-Echarri M, Rueda C, Martul P, Mayayo E, Ferrández Longás A (March 2004). "Childhood-onset congenital adrenal hyperplasia: long-term outcome and optimization of therapy". Journal of Pediatric Endocrinology & Metabolism. 17 (Suppl 3): 411–422. PMID15134301.
↑ Bachelot G, Bachelot A, Bonnier M, Salem JE, Farabos D, Trabado S, etal. (February 2023). "Combining metabolomics and machine learning models as a tool to distinguish non-classic 21-hydroxylase deficiency from polycystic ovary syndrome without adrenocorticotropic hormone testing". Human Reproduction. 38 (2): 266–276. doi:10.1093/humrep/deac254. PMID36427016.
↑ Dineen R, Martin-Grace J, Thompson CJ, Sherlock M (June 2020). "The management of glucocorticoid deficiency: Current and future perspectives". Clinica Chimica Acta; International Journal of Clinical Chemistry. 505: 148–159. doi:10.1016/j.cca.2020.03.006. PMID32145273. S2CID212629520.
↑ Karachaliou FH, Kafetzi M, Dracopoulou M, Vlachopapadopoulou E, Leka S, Fotinou A, Michalacos S (December 2016). "Cortisol response to adrenocorticotropin testing in non-classical congenital adrenal hyperplasia (NCCAH)". Journal of Pediatric Endocrinology & Metabolism. 29 (12): 1365–1371. doi:10.1515/jpem-2016-0216. PMID27849625. S2CID43390012.
↑ Klein KO, Dragnic S, Soliman AM, Bacher P (June 2018). "Predictors of bone maturation, growth rate and adult height in children with central precocious puberty treated with depot leuprolide acetate". Journal of Pediatric Endocrinology & Metabolism. 31 (6): 655–663. doi:10.1515/jpem-2017-0523. PMID29750651. S2CID21666126.
↑ Soliman AT, AlLamki M, AlSalmi I, Asfour M (May 1997). "Congenital adrenal hyperplasia complicated by central precocious puberty: linear growth during infancy and treatment with gonadotropin-releasing hormone analog". Metabolism. 46 (5): 513–517. doi:10.1016/s0026-0495(97)90186-4. PMID9160816.
↑ Halper A, Sanchez B, Hodges JS, Dengel DR, Petryk A, Sarafoglou K (July 2019). "Use of an aromatase inhibitor in children with congenital adrenal hyperplasia: Impact of anastrozole on bone mineral density and visceral adipose tissue". Clinical Endocrinology. 91 (1): 124–130. doi:10.1111/cen.14009. PMID31070802. S2CID148569761.
↑ Yang Y, Ouyang N, Ye Y, Hu Q, Du T, Di N, etal. (October 2020). "The predictive value of total testosterone alone for clinical hyperandrogenism in polycystic ovary syndrome". Reproductive Biomedicine Online. 41 (4): 734–742. doi:10.1016/j.rbmo.2020.07.013. PMID32912651. S2CID221625488.
↑ Kamrath C, Wettstaedt L, Boettcher C, Hartmann MF, Wudy SA (April 2018). "Androgen excess is due to elevated 11-oxygenated androgens in treated children with congenital adrenal hyperplasia". The Journal of Steroid Biochemistry and Molecular Biology. 178. Elsevier BV: 221–228. doi:10.1016/j.jsbmb.2017.12.016. PMID29277706. S2CID3709499.
↑ Decourt J, Jayle MF, Baulieu E (May 1957). "[Clinically late virilism with excretion of pregnanetriol and insufficiency of cortisol production]" [Clinically late virilism with excretion of pregnanetriol and insufficiency of cortisol production]. Annales d'Endocrinologie (in French). 18 (3): 416–422. PMID13470408.
↑ New MI, Lorenzen F, Pang S, Gunczler P, Dupont B, Levine LS (February 1979). ""Acquired" adrenal hyperplasia with 21-hydroxylase deficiency is not the same genetic disorders as congenital adrenal hyperplasia". The Journal of Clinical Endocrinology and Metabolism. 48 (2): 356–359. doi:10.1210/jcem-48-2-356. PMID218988.
↑ Trakakis E, Loghis C, Kassanos D (March 2009). "Congenital adrenal hyperplasia because of 21-hydroxylase deficiency. A genetic disorder of interest to obstetricians and gynecologists". Obstetrical & Gynecological Survey. 64 (3): 177–189. doi:10.1097/OGX.0b013e318193301b. PMID19228439. S2CID37242194.
↑ Concolino P (October 2019). "Issues with the Detection of Large Genomic Rearrangements in Molecular Diagnosis of 21-Hydroxylase Deficiency". Molecular Diagnosis & Therapy. 23 (5): 563–567. doi:10.1007/s40291-019-00415-z. PMID31317337. S2CID197543506.
↑ Neocleous V, Shammas C, Phedonos AP, Karaoli E, Kyriakou A, Toumba M, etal. (September 2012). "Genetic defects in the cyp21a2 gene in heterozygous girls with premature adrenarche and adolescent females with hyperandrogenemia". Georgian Medical News (210): 40–47. PMID23045419.
↑ Admoni O, Israel S, Lavi I, Gur M, Tenenbaum-Rakover Y (June 2006). "Hyperandrogenism in carriers of CYP21 mutations: the role of genotype". Clinical Endocrinology. 64 (6): 645–651. doi:10.1111/j.1365-2265.2006.02521.x. PMID16712666. S2CID37571628.
↑ Félix-López X, Riba L, Ordóñez-Sánchez ML, Ramírez-Jiménez S, Ventura-Gallegos JL, Zentella-Dehesa A, Tusié-Luna MT (September 2003). "Steroid 21-hydroxylase (P450c21) naturally occurring mutants I172N, V281L and I236n/V237E/M239K exert a dominant negative effect on enzymatic activity when co-expressed with the wild-type protein". Journal of Pediatric Endocrinology & Metabolism. 16 (7): 1017–1024. doi:10.1515/jpem.2003.16.7.1017. PMID14513879. S2CID40680772.
↑ Deaton MA, Glorioso JE, McLean DB (March 1999). "Congenital adrenal hyperplasia: not really a zebra". Am Fam Physician. 59 (5): 1190–6, 1172. PMID10088875.
↑ Forest MG, Tardy V, Nicolino M, David M, Morel Y (June 2005). "21-Hydroxylase deficiency: an exemplary model of the contribution of molecular biology in the understanding and management of the disease". Annales d'Endocrinologie. 66 (3): 225–232. doi:10.1016/s0003-4266(05)81754-8. PMID15988383.
1 2 Yoon YA, Woo S, Kim MS, Kim B, Choi YJ (April 2023). "Establishing 17-Hydroxyprogesterone Cutoff Values for Congenital Adrenal Hyperplasia in Preterm, Low Birth Weight, and Sick Newborns". Experimental and Clinical Endocrinology & Diabetes. 131 (4): 216–221. doi:10.1055/a-2022-8399. PMID36854385. S2CID257255642.
↑ Kwon C, Farrell PM (July 2000). "The magnitude and challenge of false-positive newborn screening test results". Archives of Pediatrics & Adolescent Medicine. 154 (7): 714–718. doi:10.1001/archpedi.154.7.714. PMID10891024.
↑ Therrell BL (March 2001). "Newborn screening for congenital adrenal hyperplasia". Endocrinology and Metabolism Clinics of North America. 30 (1): 15–30. doi:10.1016/s0889-8529(08)70017-3. PMID11344933.
↑ Gau M, Konishi K, Takasawa K, Nakagawa R, Tsuji-Hosokawa A, Hashimoto A, etal. (February 2021). "The progression of salt-wasting and the body weight change during the first 2 weeks of life in classical 21-hydroxylase deficiency patients". Clinical Endocrinology. 94 (2): 229–236. doi:10.1111/cen.14347. PMID33001476. S2CID222165169.
↑ Cristoni S, Cuccato D, Sciannamblo M, Bernardi LR, Biunno I, Gerthoux P, etal. (2004). "Analysis of 21-deoxycortisol, a marker of congenital adrenal hyperplasia, in blood by atmospheric pressure chemical ionization and electrospray ionization using multiple reaction monitoring". Rapid Communications in Mass Spectrometry. 18 (1): 77–82. Bibcode:2004RCMS...18...77C. doi:10.1002/rcm.1284. PMID14689562.
↑ Held PK, Bialk ER, Lasarev MR, Allen DB (March 2022). "21-Deoxycortisol is a Key Screening Marker for 21-Hydroxylase Deficiency". The Journal of Pediatrics. 242: 213–219.e1. doi:10.1016/j.jpeds.2021.10.063. PMID34780778. S2CID244106268.
↑ Gueux B, Fiet J, Galons H, Boneté R, Villette JM, Vexiau P, etal. (January 1987). "The measurement of 11 beta-hydroxy-4-pregnene-3,20-dione (21-deoxycorticosterone) by radioimmunoassay in human plasma". Journal of Steroid Biochemistry. 26 (1): 145–150. doi:10.1016/0022-4731(87)90043-4. PMID3546944.
↑ Fiet J, Gueux B, Raux-DeMay MC, Kuttenn F, Vexiau P, Brerault JL, etal. (March 1989). "Increased plasma 21-deoxycorticosterone (21-DB) levels in late-onset adrenal 21-hydroxylase deficiency suggest a mild defect of the mineralocorticoid pathway". The Journal of Clinical Endocrinology and Metabolism. 68 (3): 542–547. doi:10.1210/jcem-68-3-542. PMID2537337.
↑ D'aurizio F, Cantù M (September 2018). "Clinical endocrinology and hormones quantitation: the increasing role of mass spectrometry". Minerva Endocrinologica. 43 (3): 261–284. doi:10.23736/S0391-1977.17.02764-X. PMID29083134. S2CID12984040.
1 2 Nimkarn S, New MI (March 2010). "Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: A paradigm for prenatal diagnosis and treatment". Annals of the New York Academy of Sciences. 1192: 5–11. doi:10.1111/j.1749-6632.2009.05225.x. PMID20392211. S2CID38359933.
↑ Khattab A, Yuen T, Sun L, Yau M, Barhan A, Zaidi M, Lo YM, New MI (2016). "Noninvasive Prenatal Diagnosis of Congenital Adrenal Hyperplasia". Advanced Therapies in Pediatric Endocrinology and Diabetology. Endocrine Development. Vol.30. S.Karger AG. pp.37–41. doi:10.1159/000439326. ISBN978-3-318-05636-5. PMID26683339.
↑ Miller WL, Witchel SF (May 2013). "Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits". American Journal of Obstetrics and Gynecology. 208 (5): 354–359. doi:10.1016/j.ajog.2012.10.885. PMID23123167.
↑ Miller WL (June 2015). "Fetal endocrine therapy for congenital adrenal hyperplasia should not be done". Best Practice & Research. Clinical Endocrinology & Metabolism. 29 (3): 469–483. doi:10.1016/j.beem.2015.01.005. PMID26051303.
↑ Nordenström A, Lajic S, Falhammar H (June 2021). "Clinical outcomes in 21-hydroxylase deficiency". Current Opinion in Endocrinology, Diabetes, and Obesity. 28 (3): 318–324. doi:10.1097/MED.0000000000000625. PMID33741777. S2CID232298877.
↑ Dabas A, Vats P, Sharma R, Singh P, Seth A, Jain V, Batra P, Gupta N, Kumar R, Kabra M, Kapoor S, Yadav S (February 2020). "Management of Infants with Congenital Adrenal Hyperplasia". Indian Pediatr. 57 (2): 159–164. doi:10.1007/s13312-020-1735-8. PMID32060243. S2CID211122771.
↑ Schagen SE, Cohen-Kettenis PT, Delemarre-van de Waal HA, Hannema SE (July 2016). "Efficacy and Safety of Gonadotropin-Releasing Hormone Agonist Treatment to Suppress Puberty in Gender Dysphoric Adolescents". J Sex Med. 13 (7): 1125–32. doi:10.1016/j.jsxm.2016.05.004. PMID27318023.
↑ Szymanski KM, Rink RC, Whittam B, Hensel DJ (April 2021). "Majority of females with a life-long experience of CAH and parents do not consider females with CAH to be intersex". Journal of Pediatric Urology. 17 (2): 210.e1–210.e9. doi:10.1016/j.jpurol.2020.09.009. hdl:1805/27714. PMID33041207. S2CID222300981.
↑ Carroll L, Graff C, Wicks M, Diaz Thomas A (March 2020). "Living with an invisible illness: a qualitative study exploring the lived experiences of female children with congenital adrenal hyperplasia". Qual Life Res. 29 (3): 673–681. doi:10.1007/s11136-019-02350-2. PMID31823183. S2CID208957761.
1 2 Fausto-Sterling A (30 November 2000). Sexing the body: gender politics and the construction of sexuality. [New York]: Basic Books. ISBN9780465077137.
↑ "Intersex babies are perfect just as they are!". UN Free & Equal. up to 1.7 percent of babies are born with sex characteristics that don't fit typical definitions of male and female. That makes being intersex almost as common as being a redhead!
↑ "What is Intersex? Frequently Asked Questions". interACT: Advocates for Intersex Youth. About 1.7% of people are born intersex. (Compare that to a ~0.3% chance of having identical twins!) 1 in 2,000 babies (0.05% of humans) are born with genital differences that a doctor might suggest changing with unnecessary surgery.
↑ "Intersex population figures". Intersex Human Rights Australia. 28 September 2013. Given that intersex people only come to the attention of data collectors through chance or an apparent medical reason, the actual numbers of people with intersex variations are likely to be as much as 1.7%. Despite the limitations of the data, 1.7% seems more justifiable as an upper limit figure than alternatives, to date.
↑ Sax L (August 2002). "How common is intersex? a response to Anne Fausto-Sterling". Journal of Sex Research. 39 (3): 174–8. doi:10.1080/00224490209552139. PMID12476264. S2CID33795209. Reviewing the list of conditions which Fausto-Sterling considers to be intersex, we find that this one condition–late-onset congenital adrenal hyperplasia (LOCAH)–accounts for 88% of all those patients whom Fausto-Sterling classifies as intersex (1.5/1.7 = 88%). From a clinician's perspective, however, LOCAH is not an intersex condition. The genitalia of these babies are normal at birth, and consonant with their chromosomes: XY males have normal male genitalia, and XX females have normal female genitalia.
↑ Best J (14 September 2013). Stat-spotting: a field guide to identifying dubious data (Updated and expanded.). Berkeley: University of California Press. pp.12–13. ISBN978-0520279988.
↑ Gondim R, Teles F, Barroso U (December 2018). "Sexual orientation of 46, XX patients with congenital adrenal hyperplasia: a descriptive review". Journal of Pediatric Urology. 14 (6): 486–493. doi:10.1016/j.jpurol.2018.08.004. PMID30322770. S2CID53507149.
↑ Meyer-Bahlburg HF (March 2001). "Gender and sexuality in classic congenital adrenal hyperplasia". Endocrinology and Metabolism Clinics of North America. 30 (1): 155–71, viii. doi:10.1016/s0889-8529(08)70024-0. PMID11344934.
↑ Zainuddin AA, Grover SR, Shamsuddin K, Mahdy ZA (December 2013). "Research on quality of life in female patients with congenital adrenal hyperplasia and issues in developing nations". Journal of Pediatric and Adolescent Gynecology. 26 (6): 296–304. doi:10.1016/j.jpag.2012.08.004. PMID23507003.
↑ Finkielstain GP, Rey RA (2023). "Challenges in managing disorders of sex development associated with adrenal dysfunction". Expert Review of Endocrinology & Metabolism. 18 (5): 427–439. doi:10.1080/17446651.2023.2256393. PMID37694439. S2CID261575934.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.