
Ketosis is a metabolic state characterized by elevated levels of ketone bodies in the blood or urine. Physiological ketosis is a normal response to low glucose availability, such as low-carbohydrate diets or fasting, that provides an additional energy source for the brain in the form of ketones. In physiological ketosis, ketones in the blood are elevated above baseline levels, but the body's acid–base homeostasis is maintained. This contrasts with ketoacidosis, an uncontrolled production of ketones that occurs in pathologic states and causes a metabolic acidosis, which is a medical emergency. Ketoacidosis is most commonly the result of complete insulin deficiency in type 1 diabetes or late-stage type 2 diabetes. Ketone levels can be measured in blood, urine or breath and are generally between 0.5 and 3.0 millimolar (mM) in physiological ketosis, while ketoacidosis may cause blood concentrations greater than 10 mM.

Ketogenesis is the biochemical process through which organisms produce ketone bodies by breaking down fatty acids and ketogenic amino acids. The process supplies energy to certain organs, particularly the brain, heart and skeletal muscle, under specific scenarios including fasting, caloric restriction, sleep, or others.
Propionic acidemia, also known as propionic aciduria or propionyl-CoA carboxylase deficiency, is a rare autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia.

Medium-chain acyl-CoA dehydrogenase deficiency is a disorder of fatty acid oxidation that impairs the body's ability to break down medium-chain fatty acids into acetyl-CoA. The disorder is characterized by hypoglycemia and sudden death without timely intervention, most often brought on by periods of fasting or vomiting.

Beta-ketothiolase deficiency is a rare, autosomal recessive metabolic disorder in which the body cannot properly process the amino acid isoleucine or the products of lipid breakdown. Along with SCOT deficiency, it belongs to a group of disorders called ketone utilisation disorders.

Carnitine palmitoyltransferase I deficiency is a rare metabolic disorder that prevents the body from converting certain fats called long-chain fatty acids(LCFA) into energy, particularly during periods without food. It is caused by a mutation in CPT1A on chromosome 11.

Carnitine-acylcarnitine translocase deficiency is a rare, autosomal recessive metabolic disorder that prevents the body from converting long-chain fatty acids into energy, particularly during periods without food. Carnitine, a natural substance acquired mostly through the diet, is used by cells to process fats and produce energy. People with this disorder have a faulty enzyme that prevents long-chain fatty acids from being transported into the innermost part of the mitochondria for processing.
Glutaric acidemia type 2 is an autosomal recessive metabolic disorder that is characterised by defects in the ability of the body to use proteins and fats for energy. Incompletely processed proteins and fats can build up, leading to a dangerous chemical imbalance called acidosis. It is a metabolic myopathy, categorized under fatty acid metabolism disorder as that is the bioenergetic system that it affects the most. It also affects choline metabolism.

3-Methylcrotonyl-CoA carboxylase deficiency also known as 3-Methylcrotonylglycinuria is an inborn error of leucine metabolism and is inherited through an autosomal dominant fashion. 3-Methylcrotonyl-CoA carboxylase deficiency is caused by mutations in the MCCC1 gene, formerly known as MMCA, or the MCCC2 gene, formerly known as MCCB. MCCC1 encodes the a-subunits of 3-methylcrotonyl-CoA carboxylase while MCCC2 encodes the b-subunits. The clinical presentation of 3-Methylcrotonyl-CoA carboxylase deficiency is varied, even within members of the same family.
Pyruvate dehydrogenase deficiency is a rare neurodegenerative disorder associated with abnormal mitochondrial metabolism. PDCD is a genetic disease resulting from mutations in one of the components of the pyruvate dehydrogenase complex (PDC). The PDC is a multi-enzyme complex that plays a vital role as a key regulatory step in the central pathways of energy metabolism in the mitochondria. The disorder shows heterogeneous characteristics in both clinical presentation and biochemical abnormality.

β-Hydroxy β-methylglutaryl-CoA (HMG-CoA), also known as 3-hydroxy-3-methylglutaryl coenzyme A, is an intermediate in the mevalonate and ketogenesis pathways. It is formed from acetyl CoA and acetoacetyl CoA by HMG-CoA synthase. The research of Minor J. Coon and Bimal Kumar Bachhawat in the 1950s at University of Illinois led to its discovery.
Methylcrotonyl CoA carboxylase is a biotin-requiring enzyme located in the mitochondria. MCC uses bicarbonate as a carboxyl group source to catalyze the carboxylation of a carbon adjacent to a carbonyl group performing the fourth step in processing leucine, an essential amino acid.
3-Hydroxy-3-methylglutaryl-CoA lyase is an enzyme (EC 4.1.3.4 that in human is encoded by the HMGCL gene located on chromosome 1. It is a key enzyme in ketogenesis. It is a ketogenic enzyme in the liver that catalyzes the formation of acetoacetate from HMG-CoA within the mitochondria. It also plays a prominent role in the catabolism of the amino acid leucine.
3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare condition that prevents the body from converting certain fats to energy, particularly during fasting. Normally, through a process called fatty acid oxidation, several enzymes work in a step-wise fashion to metabolize fats and convert them to energy. People with 3-hydroxyacyl-coenzyme A dehydrogenase deficiency have inadequate levels of an enzyme required for a step that metabolizes groups of fats called medium chain fatty acids and short chain fatty acids; for this reason this disorder is sometimes called medium- and short-chain 3-hydroxyacyl-coenzyme A dehydrogenase (M/SCHAD) deficiency.

Acetoacetyl CoA is the precursor of HMG-CoA in the mevalonate pathway, which is essential for cholesterol biosynthesis. It also takes a similar role in the ketone bodies synthesis (ketogenesis) pathway of the liver. In the ketone bodies digestion pathway, it is no longer associated with having HMG-CoA as a product or as a reactant.

3-Methylglutaconyl-CoA hydratase, also known as MG-CoA hydratase and AUH, is an enzyme encoded by the AUH gene on chromosome 19. It is a member of the enoyl-CoA hydratase/isomerase superfamily, but it is the only member of that family that is able to bind to RNA. Not only does it bind to RNA, AUH has also been observed to be involved in the metabolic enzymatic activity, making it a dual-role protein. Mutations of this gene have been found to cause a disease called 3-Methylglutaconic Acuduria Type 1.

In molecular biology, hydroxymethylglutaryl-CoA synthase or HMG-CoA synthase EC 2.3.3.10 is an enzyme which catalyzes the reaction in which acetyl-CoA condenses with acetoacetyl-CoA to form 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA). This reaction comprises the second step in the mevalonate-dependent isoprenoid biosynthesis pathway. HMG-CoA is an intermediate in both cholesterol synthesis and ketogenesis. This reaction is overactivated in patients with diabetes mellitus type 1 if left untreated, due to prolonged insulin deficiency and the exhaustion of substrates for gluconeogenesis and the TCA cycle, notably oxaloacetate. This results in shunting of excess acetyl-CoA into the ketone synthesis pathway via HMG-CoA, leading to the development of diabetic ketoacidosis.
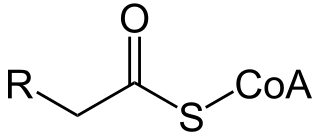
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.

3-hydroxy-3-methylglutaryl-CoA synthase 2 (mitochondrial) is an enzyme in humans that is encoded by the HMGCS2 gene.
