The urea cycle (also known as the ornithine cycle) is a cycle of biochemical reactions that produces urea (NH2)2CO from ammonia (NH3). This cycle occurs in ureotelic organisms. The urea cycle converts highly toxic ammonia to urea for excretion. This cycle was the first metabolic cycle to be discovered (Hans Krebs and Kurt Henseleit, 1932), five years before the discovery of the TCA cycle. This cycle was described in more detail later on by Ratner and Cohen. The urea cycle takes place primarily in the liver and, to a lesser extent, in the kidneys.

Arginine is the amino acid with the formula (H2N)(HN)CN(H)(CH2)3CH(NH2)CO2H. The molecule features a guanidino group appended to a standard amino acid framework. At physiological pH, the carboxylic acid is deprotonated (−CO2−) and both the amino and guanidino groups are protonated, resulting in a cation. Only the l-arginine (symbol Arg or R) enantiomer is found naturally. Arg residues are common components of proteins. It is encoded by the codons CGU, CGC, CGA, CGG, AGA, and AGG. The guanidine group in arginine is the precursor for the biosynthesis of nitric oxide. Like all amino acids, it is a white, water-soluble solid.
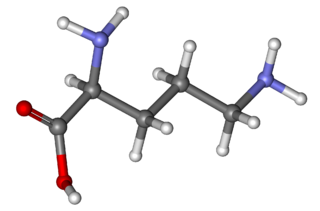
Ornithine is a non-proteinogenic amino acid that plays a role in the urea cycle. Ornithine is abnormally accumulated in the body in ornithine transcarbamylase deficiency. The radical is ornithyl.

The organic compound citrulline is an α-amino acid. Its name is derived from citrullus, the Latin word for watermelon. Although named and described by gastroenterologists since the late 19th century, it was first isolated from watermelon in 1914 by Japanese researchers Yotaro Koga and Ryo Odake and further codified by Mitsunori Wada of Tokyo Imperial University in 1930. It has the formula H2NC(O)NH(CH2)3CH(NH2)CO2H. It is a key intermediate in the urea cycle, the pathway by which mammals excrete ammonia by converting it into urea. Citrulline is also produced as a byproduct of the enzymatic production of nitric oxide from the amino acid arginine, catalyzed by nitric oxide synthase.

Ornithine transcarbamylase (OTC) is an enzyme that catalyzes the reaction between carbamoyl phosphate (CP) and ornithine (Orn) to form citrulline (Cit) and phosphate (Pi). There are two classes of OTC: anabolic and catabolic. This article focuses on anabolic OTC. Anabolic OTC facilitates the sixth step in the biosynthesis of the amino acid arginine in prokaryotes. In contrast, mammalian OTC plays an essential role in the urea cycle, the purpose of which is to capture toxic ammonia and transform it into urea, a less toxic nitrogen source, for excretion.

Hyperammonemia is a metabolic disturbance characterised by an excess of ammonia in the blood. It is a dangerous condition that may lead to brain injury and death. It may be primary or secondary.

Ornithine transcarbamylase deficiency also known as OTC deficiency is the most common urea cycle disorder in humans. Ornithine transcarbamylase, the defective enzyme in this disorder is the final enzyme in the proximal portion of the urea cycle, responsible for converting carbamoyl phosphate and ornithine into citrulline. OTC deficiency is inherited in an X-linked recessive manner, meaning males are more commonly affected than females.

N-Acetylglutamic acid (also referred to as N-acetylglutamate, abbreviated NAG, chemical formula C7H11NO5) is biosynthesized from glutamate and acetylornithine by ornithine acetyltransferase, and from glutamic acid and acetyl-CoA by the enzyme N-acetylglutamate synthase. The reverse reaction, hydrolysis of the acetyl group, is catalyzed by a specific hydrolase. It is the first intermediate involved in the biosynthesis of arginine in prokaryotes and simple eukaryotes and a regulator in the process known as the urea cycle that converts toxic ammonia to urea for excretion from the body in vertebrates.

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.

ASL is an enzyme that catalyzes the reversible breakdown of argininosuccinate (ASA) producing the amino acid arginine and dicarboxylic acid fumarate. Located in liver cytosol, ASL is the fourth enzyme of the urea cycle and involved in the biosynthesis of arginine in all species and the production of urea in ureotelic species. Mutations in ASL, resulting low activity of the enzyme, increase levels of urea in the body and result in various side effects.

Cyanophycin, also known as CGP or multi-L-arginyl-poly, is a non-protein, non-ribosomally produced amino acid polymer composed of an aspartic acid backbone and arginine side groups.

Sodium phenylbutyrate is a salt of an aromatic fatty acid, 4-phenylbutyrate (4-PBA) or 4-phenylbutyric acid. The compound is used to treat urea cycle disorders, because its metabolites offer an alternative pathway to the urea cycle to allow excretion of excess nitrogen. It is an orphan drug, marketed by Ucyclyd Pharma under the trade name Buphenyl, by Swedish Orphan International (Sweden) as Ammonaps, by Fyrlklövern Scandinavia as triButyrate and by Scandinavian Formulas, Inc..

N-Acetylglutamate synthase deficiency is an autosomal recessive urea cycle disorder.
Ornithine translocase deficiency, also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is a rare autosomal recessive urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.

Phenylacetylglutamine is a product formed by the conjugation of phenylacetate and glutamine. It is a common metabolite that occurs naturally in human urine.

Y+L amino acid transporter 1 is a protein that in humans is encoded by the SLC7A7 gene.
Delta-1-pyrroline-5-carboxylate synthetase (P5CS) is an enzyme that in humans is encoded by the ALDH18A1 gene. This gene is a member of the aldehyde dehydrogenase family and encodes a bifunctional ATP- and NADPH-dependent mitochondrial enzyme with both gamma-glutamyl kinase and gamma-glutamyl phosphate reductase activities. The encoded protein catalyzes the reduction of glutamate to delta1-pyrroline-5-carboxylate, a critical step in the de novo biosynthesis of proline, ornithine and arginine. Mutations in this gene lead to hyperammonemia, hypoornithinemia, hypocitrullinemia, hypoargininemia and hypoprolinemia and may be associated with neurodegeneration, cataracts and connective tissue diseases. Alternatively spliced transcript variants, encoding different isoforms, have been described for this gene. As reported by Bruno Reversade and colleagues, ALDH18A1 deficiency or dominant-negative mutations in P5CS in humans causes a progeroid disease known as De Barsy Syndrome.
Ornithine aminotransferase deficiency is an inborn error of ornithine metabolism, caused by decreased activity of the enzyme ornithine aminotransferase. Biochemically, it can be detected by elevated levels of ornithine in the blood. Clinically, it presents initially with poor night vision, which slowly progresses to total blindness. It is believed to be inherited in an autosomal recessive manner. Approximately 200 known cases have been reported in the literature. The incidence is highest in Finland, estimated at 1:50,000.
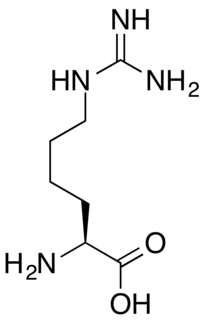
Homoarginine is an nonproteinogenic alpha-amino acid. It is structurally equivalent to a one-methylene group-higher homolog of arginine and to the guanidino derivative of lysine. L-Homoarginine is the naturally-occurring enantiomer. Physiologically, homoarginine increases nitric oxide (NO) supply and betters endothelial functions in the body, with a particular correlation and effect towards cardiovascular outcome and mortality. At physiological pH, homoarginine is cationic: the guanidino group is protonated.

L-Homocitrulline is an amino acid and a metabolite of ornithine in mammalian metabolism. The amino acid can be detected in larger amounts in the urine of individuals with urea cycle disorders. At present, it is thought that the depletion of the ornithine supply causes the accumulation of carbamyl-phosphate in the urea cycle which may be responsible for the enhanced synthesis of homocitrulline and homoarginine. Both amino acids can be detected in urine. Amino acid analysis allows for the quantitative analysis of these amino acid metabolites in biological fluids such as urine or blood.

