The urea cycle (also known as the ornithine cycle) is a cycle of biochemical reactions that produces urea (NH2)2CO from ammonia (NH3). Animals that use this cycle, mainly amphibians and mammals, are called ureotelic.

Ornithine transcarbamylase (OTC) is an enzyme that catalyzes the reaction between carbamoyl phosphate (CP) and ornithine (Orn) to form citrulline (Cit) and phosphate (Pi). There are two classes of OTC: anabolic and catabolic. This article focuses on anabolic OTC. Anabolic OTC facilitates the sixth step in the biosynthesis of the amino acid arginine in prokaryotes. In contrast, mammalian OTC plays an essential role in the urea cycle, the purpose of which is to capture toxic ammonia and transform it into urea, a less toxic nitrogen source, for excretion.

Carbamoyl phosphate is an anion of biochemical significance. In land-dwelling animals, it is an intermediary metabolite in nitrogen disposal through the urea cycle and the synthesis of pyrimidines. Its enzymatic counterpart, carbamoyl phosphate synthetase I, interacts with a class of molecules called sirtuins, NAD dependent protein deacetylases, and ATP to form carbamoyl phosphate. CP then enters the urea cycle in which it reacts with ornithine to form citrulline.

In biochemistry, a transferase is any one of a class of enzymes that catalyse the transfer of specific functional groups from one molecule to another. They are involved in hundreds of different biochemical pathways throughout biology, and are integral to some of life's most important processes.
Propionic acidemia, also known as propionic aciduria or propionyl-CoA carboxylase deficiency, is a rare autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia.

Hyperammonemia is a metabolic disturbance characterised by an excess of ammonia in the blood. It is a dangerous condition that may lead to brain injury and death. It may be primary or secondary.

Ornithine transcarbamylase deficiency also known as OTC deficiency is the most common urea cycle disorder in humans. Ornithine transcarbamylase, the defective enzyme in this disorder, is the final enzyme in the proximal portion of the urea cycle, responsible for converting carbamoyl phosphate and ornithine into citrulline. OTC deficiency is inherited in an X-linked recessive manner, meaning males are more commonly affected than females.
In molecular biology, biosynthesis is a multi-step, enzyme-catalyzed process where substrates are converted into more complex products in living organisms. In biosynthesis, simple compounds are modified, converted into other compounds, or joined to form macromolecules. This process often consists of metabolic pathways. Some of these biosynthetic pathways are located within a single cellular organelle, while others involve enzymes that are located within multiple cellular organelles. Examples of these biosynthetic pathways include the production of lipid membrane components and nucleotides. Biosynthesis is usually synonymous with anabolism.
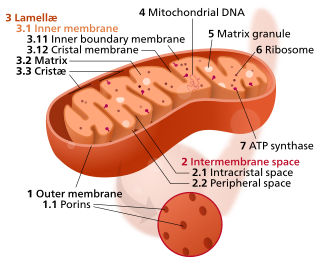
In the mitochondrion, the matrix is the space within the inner membrane. The word "matrix" stems from the fact that this space is viscous, compared to the relatively aqueous cytoplasm. The mitochondrial matrix contains the mitochondrial DNA, ribosomes, soluble enzymes, small organic molecules, nucleotide cofactors, and inorganic ions.[1] The enzymes in the matrix facilitate reactions responsible for the production of ATP, such as the citric acid cycle, oxidative phosphorylation, oxidation of pyruvate, and the beta oxidation of fatty acids.

N-Acetylglutamic acid (also referred to as N-acetylglutamate, abbreviated NAG, chemical formula C7H11NO5) is biosynthesized from glutamate and acetylornithine by ornithine acetyltransferase, and from glutamic acid and acetyl-CoA by the enzyme N-acetylglutamate synthase. The reverse reaction, hydrolysis of the acetyl group, is catalyzed by a specific hydrolase. It is the first intermediate involved in the biosynthesis of arginine in prokaryotes and simple eukaryotes and a regulator in the process known as the urea cycle that converts toxic ammonia to urea for excretion from the body in vertebrates.

The enzyme argininosuccinate lyase (EC 4.3.2.1, ASL, argininosuccinase; systematic name 2-(N ω-L-arginino)succinate arginine-lyase (fumarate-forming)) catalyzes the reversible breakdown of argininosuccinate:

Methylmalonyl-CoA mutase is a mitochondrial homodimer apoenzyme that focuses on the catalysis of methylmalonyl CoA to succinyl CoA. The enzyme is bound to adenosylcobalamin, a hormonal derivative of vitamin B12 in order to function. Methylmalonyl-CoA mutase deficiency is caused by genetic defect in the MUT gene responsible for encoding the enzyme. Deficiency in this enzyme accounts for 60% of the cases of methylmalonic acidemia.
N-Acetylglutamate synthase (NAGS) is an enzyme that catalyses the production of N-acetylglutamate (NAG) from glutamate and acetyl-CoA.
Carbamoyl phosphate synthetase I (CPS I) is a ligase enzyme located in the mitochondria involved in the production of urea. Carbamoyl phosphate synthetase I (CPS1 or CPSI) transfers an ammonia molecule to a molecule of bicarbonate that has been phosphorylated by a molecule of ATP. The resulting carbamate is then phosphorylated with another molecule of ATP. The resulting molecule of carbamoyl phosphate leaves the enzyme.
Carbamoyl phosphate synthetase I deficiency is an autosomal recessive metabolic disorder that causes ammonia to accumulate in the blood due to a lack of the enzyme carbamoyl phosphate synthetase I. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.

Carbamoyl phosphate synthetase catalyzes the ATP-dependent synthesis of carbamoyl phosphate from glutamine or ammonia and bicarbonate. This enzyme catalyzes the reaction of ATP and bicarbonate to produce carboxy phosphate and ADP. Carboxy phosphate reacts with ammonia to give carbamic acid. In turn, carbamic acid reacts with a second ATP to give carbamoyl phosphate plus ADP.
In enzymology, an aminoacylase (EC 3.5.1.14) is an enzyme that catalyzes the chemical reaction
In enzymology, a glutamate N-acetyltransferase (EC 2.3.1.35) is an enzyme that catalyzes the chemical reaction
Ornithine aminotransferase deficiency is an inborn error of ornithine metabolism, caused by decreased activity of the enzyme ornithine aminotransferase. Biochemically, it can be detected by elevated levels of ornithine in the blood. Clinically, it presents initially with poor night vision, which slowly progresses to total blindness. It is believed to be inherited in an autosomal recessive manner. Approximately 200 known cases have been reported in the literature. The incidence is highest in Finland, estimated at 1:50,000.

Citrullinemia type I (CTLN1), also known as arginosuccinate synthetase deficiency, is a rare disease caused by a deficiency in argininosuccinate synthetase, an enzyme involved in excreting excess nitrogen from the body. There are mild and severe forms of the disease, which is one of the urea cycle disorders.


