
Anemia or anaemia is a blood disorder in which the blood has a reduced ability to carry oxygen due to a lower than normal number of red blood cells, a reduction in the amount of hemoglobin or hemoglobin abnormalities. The name is derived from Ancient Greek: ἀναιμία anaimia, meaning 'lack of blood', from ἀν- an-, 'not' and αἷμα haima, 'blood'. When anemia comes on slowly, the symptoms are often vague, such as tiredness, weakness, shortness of breath, headaches, and a reduced ability to exercise. When anemia is acute, symptoms may include confusion, feeling like one is going to pass out, loss of consciousness, and increased thirst. Anemia must be significant before a person becomes noticeably pale. Symptoms of anemia depend on how quickly hemoglobin decreases. Additional symptoms may occur depending on the underlying cause. Preoperative anemia can increase the risk of needing a blood transfusion following surgery. Anemia can be temporary or long term and can range from mild to severe.

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Thalassemias are inherited blood disorders that result in abnormal hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Symptoms and presentations of thalassemia can change over time.
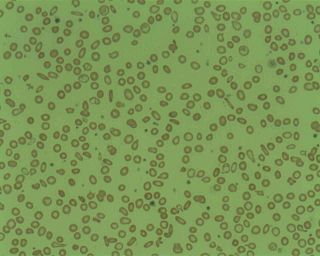
Microcytic anaemia is any of several types of anemia characterized by smaller than normal red blood cells. The normal mean corpuscular volume is approximately 80–100 fL. When the MCV is <80 fL, the red cells are described as microcytic and when >100 fL, macrocytic. The MCV is the average red blood cell size.
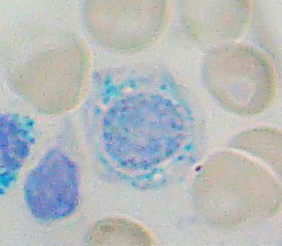
Sideroblastic anemia, or sideroachrestic anemia, is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes). In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome, which can develop into hematological malignancies.

Hypochondroplasia (HCH) is a developmental disorder caused by an autosomal dominant genetic defect in the fibroblast growth factor receptor 3 gene (FGFR3) that results in a disproportionately short stature, micromelia and a head that appears large in comparison with the underdeveloped portions of the body. It is classified as short-limbed dwarfism.

Alpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.

Oculocerebrorenal syndrome is a rare X-linked recessive disorder characterized by congenital cataracts, hypotonia, intellectual disability, proximal tubular acidosis, aminoaciduria and low-molecular-weight proteinuria. Lowe syndrome can be considered a cause of Fanconi syndrome.
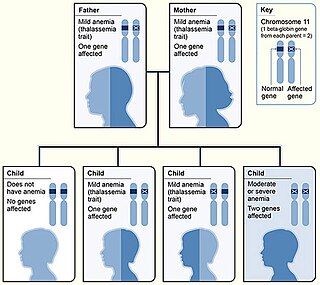
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.

Atransferrinemia is an autosomal recessive metabolic disorder in which there is an absence of transferrin, a plasma protein that transports iron through the blood. Atransferrinemia is characterized by anemia and hemosiderosis in the heart and liver. The iron damage to the heart can lead to heart failure. The anemia is typically microcytic and hypochromic. Atransferrinemia was first described in 1961 and is extremely rare, with only ten documented cases worldwide.

Hemosiderosis is a form of iron overload disorder resulting in the accumulation of hemosiderin.
Haemochromatosis type 3 is a type of iron overload disorder associated with deficiencies in transferrin receptor 2. It exhibits an autosomal recessive inheritance pattern. The first confirmed case was diagnosed in 1865 by French doctor Trousseau. Later in 1889, the German doctor von Recklinghausen indicated that the liver contains iron, and due to bleeding being considered to be the cause, he called the pigment "Haemochromatosis." In 1935, English doctor Sheldon's groundbreaking book titled, Haemochromatosis, reviewed 311 patient case reports and presented the idea that haemochromatosis was a congenital metabolic disorder. Hereditary haemochromatosis is a congenital disorder which affects the regulation of iron metabolism thus causing increased gut absorption of iron and a gradual build-up of pathologic iron deposits in the liver and other internal organs, joint capsules and the skin. The iron overload could potentially cause serious disease from the age of 40–50 years. In the final stages of the disease, the major symptoms include liver cirrhosis, diabetes and bronze-colored skin. There are four types of hereditary hemochromatosis which are classified depending on the age of onset and other factors such as genetic cause and mode of inheritance.

Ullrich congenital muscular dystrophy (UCMD) is a form of congenital muscular dystrophy. There are two forms: UCMD1 and UCMD2.
Majeed syndrome is an inherited skin disorder characterized by chronic recurrent multifocal osteomyelitis, congenital dyserythropoietic anemia and a neutrophilic dermatosis. It is classified as an autoinflammatory bone disorder. The condition is found in people with two defective copies of the LPIN2 gene. LPIN2 encodes lipin-2 which is involved in lipid metabolism. The pathogenesis of this mutation with the clinical manifestations has not been elucidated.
Congenital dyserythropoietic anemia type I is a disorder of blood cell production, particularly of the production of erythroblasts, which are the precursors of the red blood cells (RBCs).
Congenital dyserythropoietic anemia type II, or hereditary erythroblastic multinuclearity with positive acidified serum lysis test (HEMPAS) is a rare genetic anemia in humans characterized by hereditary erythroblastic multinuclearity with positive acidified serum lysis test.
Congenital dyserythropoietic anemia type III is a rare autosomal dominant disorder characterized by macrocytic anemia, bone marrow erythroid hyperplasia and giant multinucleate erythroblasts. New evidence suggests that this may be passed on recessively as well.
Congenital dyserythropoietic anemia type IV has been described with typical morphologic features of CDA II but a negative acidified-serum test.

Transfusion-dependent anemia is a form of anemia characterized by the need for continuous blood transfusion. It is a condition that results from various diseases, and is associated with decreased survival rates. Regular transfusion is required to reduce the symptoms of anemia by increasing functional red blood cells and hemoglobin count. Symptoms may vary based on the severity of the condition and the most common symptom is fatigue. Various diseases can lead to transfusion-dependent anemia, most notably myelodysplastic syndromes (MDS) and thalassemia. Due to the number of diseases that can cause transfusion-dependent anemia, diagnosing it is more complicated. Transfusion dependence occurs when an average of more than 2 units of blood transfused every 28 days is required over a period of at least 3 months. Myelodysplastic syndromes is often only diagnosed when patients become anemic, and transfusion-dependent thalassemia is diagnosed based on gene mutations. Screening for heterozygosity in the thalassemia gene is an option for early detection.
Hemolytic jaundice, also known as prehepatic jaundice, is a type of jaundice arising from hemolysis or excessive destruction of red blood cells, when the byproduct bilirubin is not excreted by the hepatic cells quickly enough. Unless the patient is concurrently affected by hepatic dysfunctions or is experiencing hepatocellular damage, the liver does not contribute to this type of jaundice.


