Related Research Articles
In electrochemistry, electrode potential is the voltage of a galvanic cell built from a standard reference electrode and another electrode to be characterized. By convention, the reference electrode is the standard hydrogen electrode (SHE). It is defined to have a potential of zero volts. It may also be defined as the potential difference between the charged metallic rods and salt solution.

In electrochemistry, cyclic voltammetry (CV) is a type of potentiodynamic measurement. In a cyclic voltammetry experiment, the working electrode potential is ramped linearly versus time. Unlike in linear sweep voltammetry, after the set potential is reached in a CV experiment, the working electrode's potential is ramped in the opposite direction to return to the initial potential. These cycles of ramps in potential may be repeated as many times as needed. The current at the working electrode is plotted versus the applied voltage to give the cyclic voltammogram trace. Cyclic voltammetry is generally used to study the electrochemical properties of an analyte in solution or of a molecule that is adsorbed onto the electrode.
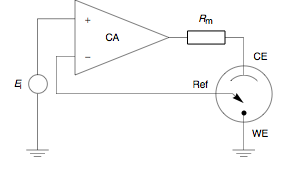
A potentiostat is the electronic hardware required to control a three electrode cell and run most electroanalytical experiments. A Bipotentiostat and polypotentiostat are potentiostats capable of controlling two working electrodes and more than two working electrodes, respectively.

Voltammetry is a category of electroanalytical methods used in analytical chemistry and various industrial processes. In voltammetry, information about an analyte is obtained by measuring the current as the potential is varied. The analytical data for a voltammetric experiment comes in the form of a voltammogram, which plots the current produced by the analyte versus the potential of the working electrode.
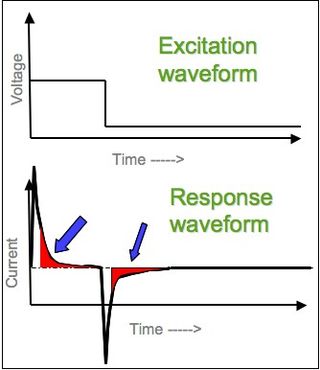
In electrochemistry, chronoamperometry is an analytical technique in which the electric potential of the working electrode is stepped and the resulting current from faradaic processes occurring at the electrode is monitored as a function of time. The functional relationship between current response and time is measured after applying single or double potential step to the working electrode of the electrochemical system. Limited information about the identity of the electrolyzed species can be obtained from the ratio of the peak oxidation current versus the peak reduction current. However, as with all pulsed techniques, chronoamperometry generates high charging currents, which decay exponentially with time as any RC circuit. The Faradaic current - which is due to electron transfer events and is most often the current component of interest - decays as described in the Cottrell equation. In most electrochemical cells, this decay is much slower than the charging decay-cells with no supporting electrolyte are notable exceptions. Most commonly a three-electrode system is used. Since the current is integrated over relatively longer time intervals, chronoamperometry gives a better signal-to-noise ratio in comparison to other amperometric techniques.
Polarography is a type of voltammetry where the working electrode is a dropping mercury electrode (DME) or a static mercury drop electrode (SMDE), which are useful for their wide cathodic ranges and renewable surfaces. It was invented in 1922 by Czechoslovak chemist Jaroslav Heyrovský, for which he won the Nobel prize in 1959. The main advantages of mercury as electrode material are as follows: 1) a large voltage window: ca. from +0.2 V to -1.8 V vs reversible hydrogen electrode (RHE). Hg electrode is particularly well-suited for studying electroreduction reactions. 2) very reproducible electrode surface, since mercury is liquid. 3) very easy cleaning of the electrode surface by making a new drop of mercury from a large Hg pool connected by a glass capillary.
In electrochemistry, overpotential is the potential difference (voltage) between a half-reaction's thermodynamically-determined reduction potential and the potential at which the redox event is experimentally observed. The term is directly related to a cell's voltage efficiency. In an electrolytic cell the existence of overpotential implies that the cell requires more energy than thermodynamically expected to drive a reaction. In a galvanic cell the existence of overpotential means less energy is recovered than thermodynamics predicts. In each case the extra/missing energy is lost as heat. The quantity of overpotential is specific to each cell design and varies across cells and operational conditions, even for the same reaction. Overpotential is experimentally determined by measuring the potential at which a given current density is achieved.
In analytical chemistry, potentiometric titration is a technique similar to direct titration of a redox reaction. It is a useful means of characterizing an acid. No indicator is used; instead the electric potential is measured across the analyte, typically an electrolyte solution. To do this, two electrodes are used, an indicator electrode and a reference electrode. Reference electrodes generally used are hydrogen electrodes, calomel electrodes, and silver chloride electrodes. The indicator electrode forms an electrochemical half-cell with the interested ions in the test solution. The reference electrode forms the other half-cell.
Electroanalytical methods are a class of techniques in analytical chemistry which study an analyte by measuring the potential (volts) and/or current (amperes) in an electrochemical cell containing the analyte. These methods can be broken down into several categories depending on which aspects of the cell are controlled and which are measured. The four main categories are potentiometry, amperometry, coulometry, and voltammetry.
In analytical chemistry, linear sweep voltammetry is a method of voltammetry where the current at a working electrode is measured while the potential between the working electrode and a reference electrode is swept linearly in time. Oxidation or reduction of species is registered as a peak or trough in the current signal at the potential at which the species begins to be oxidized or reduced.
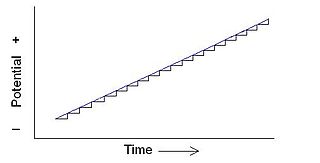
Staircase voltammetry is a derivative of linear sweep voltammetry. In linear sweep voltammetry the current at a working electrode is measured while the potential between the working electrode and a reference electrode is swept linearly in time. Oxidation or reduction of species is registered as a peak or trough in the current signal at the potential at which the species begins to be oxidized or reduced.
Squarewave voltammetry (SWV) is a form of linear potential sweep voltammetry that uses a combined square wave and staircase potential applied to a stationary electrode. It has found numerous applications in various fields, including within medicinal and various sensing communities.
In electrochemistry, the auxiliary electrode, often also called the counter electrode, is an electrode used in a three-electrode electrochemical cell for voltammetric analysis or other reactions in which an electric current is expected to flow. The auxiliary electrode is distinct from the reference electrode, which establishes the electrical potential against which other potentials may be measured, and the working electrode, at which the cell reaction takes place.
An ultramicroelectrode (UME) is a working electrode used in a voltammetry. The small size of UME give them large diffusion layers and small overall currents. These features allow UME to achieve useful steady-state conditions and very high scan rates (V/s) with limited distortion. UME were developed independently by Wightman and Fleischmann around 1980. Small current at UME enables electrochemical measurements in low conductive media, where voltage drop associated with high solution resistance makes these experiments difficult for conventional electrodes. Furthermore, small voltage drop at UME leads to a very small voltage distortion at the electrode-solution interface which allows using two-electrode setup in voltammetric experiment instead of conventional three-electrode setup.
In analytical chemistry, hydrodynamic voltammetry is a form of voltammetry in which the analyte solution flows relative to a working electrode. In many voltammetry techniques, the solution is intentionally left still to allow diffusion-controlled mass transfer. When a solution is made to flow, through stirring or some other physical mechanism, it is very important to the technique to achieve a very controlled flux or mass transfer in order to obtain predictable results. These methods are types of electrochemical studies which use potentiostats to investigate reaction mechanisms related to redox chemistry among other chemical phenomenon.
In electrochemistry, the Randles–Ševčík equation describes the effect of scan rate on the peak current ip for a cyclic voltammetry experiment. For simple redox events such as the ferrocene/ferrocenium couple, ip depends not only on the concentration and diffusional properties of the electroactive species but also on scan rate.
A liquid metal electrode is an electrode that uses a liquid metal, such as mercury, Galinstan, and NaK. They can be used in electrocapillarity, voltammetry, and impedance measurements.

Fast-scan cyclic voltammetry (FSCV) is cyclic voltammetry with a very high scan rate (up to 1×106 V·s−1). Application of high scan rate allows rapid acquisition of a voltammogram within several milliseconds and ensures high temporal resolution of this electroanalytical technique. An acquisition rate of 10 Hz is routinely employed.
In electrochemistry, protein film voltammetry is a technique for examining the behavior of proteins immobilized on an electrode. The technique is applicable to proteins and enzymes that engage in electron transfer reactions and it is part of the methods available to study enzyme kinetics.

Electrochemical stripping analysis is a set of analytical chemistry methods based on voltammetry or potentiometry that are used for quantitative determination of ions in solution. Stripping voltammetry have been employed for analysis of organic molecules as well as metal ions. Carbon paste, glassy carbon paste, and glassy carbon electrodes when modified are termed as chemically modified electrodes and have been employed for the analysis of organic and inorganic compounds.
References
- ↑ Fritz Scholz (21 December 2013). Electroanalytical Methods: Guide to Experiments and Applications. Springer. pp. 109–. ISBN 978-3-662-04757-6.
- ↑ Laborda, Eduardo; González, Joaquín; Molina, Ángela (2014). "Recent advances on the theory of pulse techniques: A mini review". Electrochemistry Communications. 43: 25–30. doi:10.1016/j.elecom.2014.03.004. ISSN 1388-2481.
- ↑ García-Armada, Pilar; Losada, José; de Vicente-Pérez, Santiago (1996). "Cation Analysis Scheme by Differential Pulse Polarography". Journal of Chemical Education. 73 (6): 544. Bibcode:1996JChEd..73..544G. doi:10.1021/ed073p544. ISSN 0021-9584.