Metagenomics is the study of genetic material recovered directly from environmental or clinical samples by a method called sequencing. The broad field may also be referred to as environmental genomics, ecogenomics, community genomics or microbiomics.

16S ribosomal RNA is the RNA component of the 30S subunit of a prokaryotic ribosome. It binds to the Shine-Dalgarno sequence and provides most of the SSU structure.

The Human Microbiome Project (HMP) was a United States National Institutes of Health (NIH) research initiative to improve understanding of the microbiota involved in human health and disease. Launched in 2007, the first phase (HMP1) focused on identifying and characterizing human microbiota. The second phase, known as the Integrative Human Microbiome Project (iHMP) launched in 2014 with the aim of generating resources to characterize the microbiome and elucidating the roles of microbes in health and disease states. The program received $170 million in funding by the NIH Common Fund from 2007 to 2016.

Microbiota are the range of microorganisms that may be commensal, mutualistic, or pathogenic found in and on all multicellular organisms, including plants. Microbiota include bacteria, archaea, protists, fungi, and viruses, and have been found to be crucial for immunologic, hormonal, and metabolic homeostasis of their host.
Community fingerprinting is a set of molecular biology techniques that can be used to quickly profile the diversity of a microbial community. Rather than directly identifying or counting individual cells in an environmental sample, these techniques show how many variants of a gene are present. In general, it is assumed that each different gene variant represents a different type of microbe. Community fingerprinting is used by microbiologists studying a variety of microbial systems to measure biodiversity or track changes in community structure over time. The method analyzes environmental samples by assaying genomic DNA. This approach offers an alternative to microbial culturing, which is important because most microbes cannot be cultured in the laboratory. Community fingerprinting does not result in identification of individual microbe species; instead, it presents an overall picture of a microbial community. These methods are now largely being replaced by high throughput sequencing, such as targeted microbiome analysis and metagenomics.
Biological dark matter is an informal term for unclassified or poorly understood genetic material. This genetic material may refer to genetic material produced by unclassified microorganisms. By extension, biological dark matter may also refer to the un-isolated microorganisms whose existence can only be inferred from the genetic material that they produce. Some of the genetic material may not fall under the three existing domains of life: Bacteria, Archaea and Eukaryota; thus, it has been suggested that a possible fourth domain of life may yet be discovered, although other explanations are also probable. Alternatively, the genetic material may refer to non-coding DNA and non-coding RNA produced by known organisms.
Microbial phylogenetics is the study of the manner in which various groups of microorganisms are genetically related. This helps to trace their evolution. To study these relationships biologists rely on comparative genomics, as physiology and comparative anatomy are not possible methods.

Viral metagenomics uses metagenomic technologies to detect viral genomic material from diverse environmental and clinical samples. Viruses are the most abundant biological entity and are extremely diverse; however, only a small fraction of viruses have been sequenced and only an even smaller fraction have been isolated and cultured. Sequencing viruses can be challenging because viruses lack a universally conserved marker gene so gene-based approaches are limited. Metagenomics can be used to study and analyze unculturable viruses and has been an important tool in understanding viral diversity and abundance and in the discovery of novel viruses. For example, metagenomics methods have been used to describe viruses associated with cancerous tumors and in terrestrial ecosystems.
Single-cell sequencing examines the nucleic acid sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.
Mark J. Pallen is a research leader at the Quadram Institute and Professor of Microbial Genomics at the University of East Anglia. In recent years, he has been at the forefront of efforts to apply next-generation sequencing to problems in microbiology and ancient DNA research.

A microbiome is the community of microorganisms that can usually be found living together in any given habitat. It was defined more precisely in 1988 by Whipps et al. as "a characteristic microbial community occupying a reasonably well-defined habitat which has distinct physio-chemical properties. The term thus not only refers to the microorganisms involved but also encompasses their theatre of activity". In 2020, an international panel of experts published the outcome of their discussions on the definition of the microbiome. They proposed a definition of the microbiome based on a revival of the "compact, clear, and comprehensive description of the term" as originally provided by Whipps et al., but supplemented with two explanatory paragraphs, the first pronouncing the dynamic character of the microbiome, and the second clearly separating the term microbiota from the term microbiome.
Microbial dark matter (MDM) comprises the vast majority of microbial organisms that microbiologists are unable to culture in the laboratory, due to lack of knowledge or ability to supply the required growth conditions. Microbial dark matter is analogous to the dark matter of physics and cosmology due to its elusiveness in research and importance to our understanding of biological diversity. Microbial dark matter can be found ubiquitously and abundantly across multiple ecosystems, but remains difficult to study due to difficulties in detecting and culturing these species, posing challenges to research efforts. It is difficult to estimate its relative magnitude, but the accepted gross estimate is that as little as one percent of microbial species in a given ecological niche are culturable. In recent years, more effort has been directed towards deciphering microbial dark matter by means of recovering genome DNA sequences from environmental samples via culture independent methods such as single cell genomics and metagenomics. These studies have enabled insights into the evolutionary history and the metabolism of the sequenced genomes, providing valuable knowledge required for the cultivation of microbial dark matter lineages. However, microbial dark matter research remains comparatively undeveloped and is hypothesized to provide insight into processes radically different from known biology, new understandings of microbial communities, and increasing understanding of how life survives in extreme environments.
Metatranscriptomics is the set of techniques used to study gene expression of microbes within natural environments, i.e., the metatranscriptome.
PICRUSt is a bioinformatics software package. The name is an abbreviation for Phylogenetic Investigation of Communities by Reconstruction of Unobserved States.
Virome refers to the assemblage of viruses that is often investigated and described by metagenomic sequencing of viral nucleic acids that are found associated with a particular ecosystem, organism or holobiont. The word is frequently used to describe environmental viral shotgun metagenomes. Viruses, including bacteriophages, are found in all environments, and studies of the virome have provided insights into nutrient cycling, development of immunity, and a major source of genes through lysogenic conversion. Also, the human virome has been characterized in nine organs of 31 Finnish individuals using qPCR and NGS methodologies.
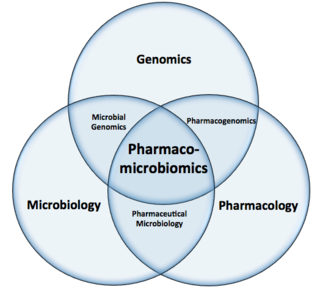
Pharmacomicrobiomics, proposed by Prof. Marco Candela for the ERC-2009-StG project call, and publicly coined for the first time in 2010 by Rizkallah et al., is defined as the effect of microbiome variations on drug disposition, action, and toxicity. Pharmacomicrobiomics is concerned with the interaction between xenobiotics, or foreign compounds, and the gut microbiome. It is estimated that over 100 trillion prokaryotes representing more than 1000 species reside in the gut. Within the gut, microbes help modulate developmental, immunological and nutrition host functions. The aggregate genome of microbes extends the metabolic capabilities of humans, allowing them to capture nutrients from diverse sources. Namely, through the secretion of enzymes that assist in the metabolism of chemicals foreign to the body, modification of liver and intestinal enzymes, and modulation of the expression of human metabolic genes, microbes can significantly impact the ingestion of xenobiotics.
Machine learning in bioinformatics is the application of machine learning algorithms to bioinformatics, including genomics, proteomics, microarrays, systems biology, evolution, and text mining.
Nikos Kyrpides is a Greek-American bioscientist who has worked on the origins of life, information processing, bioinformatics, microbiology, metagenomics and microbiome data science. He is a senior staff scientist at the Berkeley National Laboratory, head of the Prokaryote Super Program and leads the Microbiome Data Science program at the US Department of Energy Joint Genome Institute.
Clinical metagenomic next-generation sequencing (mNGS) is the comprehensive analysis of microbial and host genetic material in clinical samples from patients by next-generation sequencing. It uses the techniques of metagenomics to identify and characterize the genome of bacteria, fungi, parasites, and viruses without the need for a prior knowledge of a specific pathogen directly from clinical specimens. The capacity to detect all the potential pathogens in a sample makes metagenomic next generation sequencing a potent tool in the diagnosis of infectious disease especially when other more directed assays, such as PCR, fail. Its limitations include clinical utility, laboratory validity, sense and sensitivity, cost and regulatory considerations.

A microbiome-wide association study (MWAS), otherwise known as a metagenome-wide association study (MGWAS), is a statistical methodology used to examine the full metagenome of a defined microbiome in various organisms to determine if some feature of the microbiome is associated with a host trait. MWAS has been adopted by the field of metagenomics from the widely used genome-wide association study (GWAS).