
Epilepsy is a group of non-communicable neurological disorders characterized by recurrent epileptic seizures. An epileptic seizure is the clinical manifestation of an abnormal, excessive, and synchronized electrical discharge in the brain cells called neurons. The occurrence of two or more unprovoked seizures defines epilepsy. The occurrence of just one seizure may warrant the definition in a more clinical usage where recurrence may be able to be prejudged. Epileptic seizures can vary from brief and nearly undetectable periods to long periods of vigorous shaking due to abnormal electrical activity in the brain. These episodes can result in physical injuries, either directly such as broken bones or through causing accidents. In epilepsy, seizures tend to recur and may have no immediate underlying cause. Isolated seizures that are provoked by a specific cause such as poisoning are not deemed to represent epilepsy. People with epilepsy may be treated differently in various areas of the world and experience varying degrees of social stigma due to the alarming nature of their symptoms.
Lennox–Gastaut syndrome (LGS) is a complex, rare, and severe childhood-onset epilepsy syndrome. It is characterized by multiple and concurrent seizure types including tonic seizure, cognitive dysfunction, and slow spike waves on electroencephalogram (EEG), which are very abnormal. Typically, it presents in children aged 3–5 years and most of the time persists into adulthood with slight changes in the electroclinical phenotype. It has been associated with perinatal injuries, congenital infections, brain malformations, brain tumors, genetic disorders such as tuberous sclerosis and numerous gene mutations. Sometimes LGS is observed after infantile epileptic spasm syndrome. The prognosis for LGS is marked by a 5% mortality in childhood and persistent seizures into adulthood.
Generalized epilepsy with febrile seizures plus (GEFS+) is a syndromic autosomal dominant disorder where affected individuals can exhibit numerous epilepsy phenotypes. GEFS+ can persist beyond early childhood. GEFS+ is also now believed to encompass three other epilepsy disorders: severe myoclonic epilepsy of infancy (SMEI), which is also known as Dravet's syndrome, borderline SMEI (SMEB), and intractable epilepsy of childhood (IEC). There are at least six types of GEFS+, delineated by their causative gene. Known causative gene mutations are in the sodium channel α subunit genes SCN1A, an associated β subunit SCN1B, and in a GABAA receptor γ subunit gene, in GABRG2 and there is another gene related with calcium channel the PCDH19 which is also known as Epilepsy Female with Mental Retardation. Penetrance for this disorder is estimated at 60%.
Dravet syndrome (DS), previously known as severe myoclonic epilepsy of infancy (SMEI), is an autosomal dominant genetic disorder which causes a catastrophic form of epilepsy, with prolonged seizures that are often triggered by hot temperatures or fever. It is very difficult to treat with anticonvulsant medications. It often begins before one year of age, with six months being the age that seizures, characterized by prolonged convulsions and triggered by fever, usually begin.
Childhood absence epilepsy (CAE), formerly known as pyknolepsy, is an idiopathic generalized epilepsy which occurs in otherwise normal children. The age of onset is between 4–10 years with peak age between 5–7 years. Children have absence seizures which although brief, they occur frequently, sometimes in the hundreds per day. The absence seizures of CAE involve abrupt and severe impairment of consciousness. Mild automatisms are frequent, but major motor involvement early in the course excludes this diagnosis. The EEG demonstrates characteristic "typical 3Hz spike-wave" discharges. The presence of any other seizure type at time of diagnosis rules out the diagnose of CAE. Prognosis is usually good in well-defined cases of CAE with most patients "growing out" of their epilepsy.
Idiopathic generalized epilepsy (IGE) is a group of epileptic disorders that are believed to have a strong underlying genetic basis. IGE is considered a subgroup of Genetic Generalized Epilepsy (GGE). Patients with an IGE subtype are typically otherwise normal and have no structural brain abnormalities. People also often have a family history of epilepsy and seem to have a genetically predisposed risk of seizures. IGE tends to manifest itself between early childhood and adolescence although it can be eventually diagnosed later. The genetic cause of some IGE types is known, though inheritance does not always follow a simple monogenic mechanism.

CDKL5 is a gene that provides instructions for making a protein called cyclin-dependent kinase-like 5 also known as serine/threonine kinase 9 (STK9) that is essential for normal brain development. Mutations in the gene can cause deficiencies in the protein. The gene regulates neuronal morphology through cytoplasmic signaling and controlling gene expression. The CDKL5 protein acts as a kinase, which is an enzyme that changes the activity of other proteins by adding a cluster of oxygen and phosphorus atoms at specific positions. Researchers are currently working to determine which proteins are targeted by the CDKL5 protein.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).
Ohtahara syndrome (OS), also known as early infantile epileptic encephalopathy (EIEE) is a progressive epileptic encephalopathy. The syndrome is outwardly characterized by tonic spasms and partial seizures within the first few months of life, and receives its more elaborate name from the pattern of burst activity on an electroencephalogram (EEG). It is an extremely debilitating progressive neurological disorder, involving intractable seizures and severe intellectual disabilities. No single cause has been identified, although in many cases structural brain damage is present.

Sodium channel protein type 1 subunit alpha (SCN1A), is a protein which in humans is encoded by the SCN1A gene.
Spike-and-wave is a pattern of the electroencephalogram (EEG) typically observed during epileptic seizures. A spike-and-wave discharge is a regular, symmetrical, generalized EEG pattern seen particularly during absence epilepsy, also known as ‘petit mal’ epilepsy. The basic mechanisms underlying these patterns are complex and involve part of the cerebral cortex, the thalamocortical network, and intrinsic neuronal mechanisms.
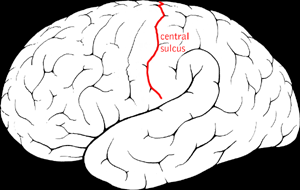
Benign Rolandic epilepsy or self-limited epilepsy with centrotemporal spikes is the most common epilepsy syndrome in childhood. Most children will outgrow the syndrome, hence the label benign. The seizures, sometimes referred to as sylvian seizures, start around the central sulcus of the brain.
Early myoclonic encephalopathy (EME) is a rare neonatal-onset epilepsy developmental and epileptic encephalopathy (DEE) with an onset at neonatal period or during the first 3 months of life. This syndrome is now included as part of the Early infantile developmental and epileptic encephalopathy (EIDEE) under the 2022 ILAE syndrome classification.
Febrile infection-related epilepsy syndrome (FIRES), is onset of severe seizures following a febrile illness in someone who was previously healthy. The seizures may initially be focal; however, often become tonic-clonic. Complications often include intellectual disability, behavioral problems, and ongoing seizures.
People with epilepsy may be classified into different syndromes based on specific clinical features. These features include the age at which seizures begin, the seizure types, and EEG findings, among others. Identifying an epilepsy syndrome is useful as it helps determine the underlying causes as well as deciding what anti-seizure medication should be tried. Epilepsy syndromes are more commonly diagnosed in infants and children. Some examples of epilepsy syndromes include benign rolandic epilepsy, childhood absence epilepsy and juvenile myoclonic epilepsy. Severe syndromes with diffuse brain dysfunction caused, at least partly, by some aspect of epilepsy, are also referred to as epileptic encephalopathies. These are associated with frequent seizures that are resistant to treatment and severe cognitive dysfunction, for instance Lennox-Gastaut syndrome and West syndrome.
Lori L. Isom is an American pharmacologist, an elected Fellow of the American Association for the Advancement of Science, and a member of the National Academy of Medicine.
SYNGAP1-related intellectual disability is a monogenetic developmental and epileptic encephalopathy that affects the central nervous system. Symptoms include intellectual disability, epilepsy, autism, sensory processing deficits, hypotonia and unstable gait.
CDKL5 deficiency disorder (CDD) is a rare genetic disorder caused by pathogenic variants in the gene CDKL5.
Malignant migrating partial seizures of infancy (MMPSI) is a rare epileptic syndrome that onsets before 6 months of age, commonly in the first few weeks of life. Once seizures start, the site of seizure activity repeatedly migrates from one area of the brain to another, with few periods of remission in between. These seizures are 'focal' (updated term for 'partial'), meaning they do not affect both sides of the brain at the same time. These continuous seizures cause damage to the brain, hence the descriptor 'malignant.'
SLC6A1 epileptic encephalopathy is a genetic disorder characterised by the loss-of-function of one copy of the human SLC6A1 gene. SLC6A1 epileptic encephalopathy can typically manifest itself with early onset seizures and it can also be characterised by mild to severe learning disability. Not all manifestations of the conditions are present in one given patient.
