
The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. A synthetic primer may also be referred to as an oligo, short for oligonucleotide. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. DNA polymerase adds nucleotides after binding to the RNA primer and synthesizes the whole strand. Later, the RNA strands must be removed accurately and replace them with DNA nucleotides forming a gap region known as a nick that is filled in using an enzyme called ligase. The removal process of the RNA primer requires several enzymes, such as Fen1, Lig1, and others that work in coordination with DNA polymerase, to ensure the removal of the RNA nucleotides and the addition of DNA nucleotides. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable. Primers can be designed in laboratory for specific reactions such as polymerase chain reaction (PCR). When designing PCR primers, there are specific measures that must be taken into consideration, like the melting temperature of the primers and the annealing temperature of the reaction itself. Moreover, the DNA binding sequence of the primer in vitro has to be specifically chosen, which is done using a method called basic local alignment search tool (BLAST) that scans the DNA and finds specific and unique regions for the primer to bind.
Protein engineering is the process of developing useful or valuable proteins through the design and production of unnatural polypeptides, often by altering amino acid sequences found in nature. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional mutating changes to the DNA sequence of a gene and any gene products. Also called site-specific mutagenesis or oligonucleotide-directed mutagenesis, it is used for investigating the structure and biological activity of DNA, RNA, and protein molecules, and for protein engineering.
In molecular biology, an amplicon is a piece of DNA or RNA that is the source and/or product of amplification or replication events. It can be formed artificially, using various methods including polymerase chain reactions (PCR) or ligase chain reactions (LCR), or naturally through gene duplication. In this context, amplification refers to the production of one or more copies of a genetic fragment or target sequence, specifically the amplicon. As it refers to the product of an amplification reaction, amplicon is used interchangeably with common laboratory terms, such as "PCR product."

Random amplified polymorphic DNA (RAPD), pronounced "rapid", is a type of polymerase chain reaction (PCR), but the segments of DNA that are amplified are random. The scientist performing RAPD creates several arbitrary, short primers, then proceeds with the PCR using a large template of genomic DNA, hoping that fragments will amplify. By resolving the resulting patterns, a semi-unique profile can be gleaned from an RAPD reaction.

In molecular biology, subcloning is a technique used to move a particular DNA sequence from a parent vector to a destination vector.

AFLP-PCR or just AFLP is a PCR-based tool used in genetics research, DNA fingerprinting, and in the practice of genetic engineering. Developed in the early 1990s by KeyGene, AFLP uses restriction enzymes to digest genomic DNA, followed by ligation of adaptors to the sticky ends of the restriction fragments. A subset of the restriction fragments is then selected to be amplified. This selection is achieved by using primers complementary to the adaptor sequence, the restriction site sequence and a few nucleotides inside the restriction site fragments. The amplified fragments are separated and visualized on denaturing on agarose gel electrophoresis, either through autoradiography or fluorescence methodologies, or via automated capillary sequencing instruments.
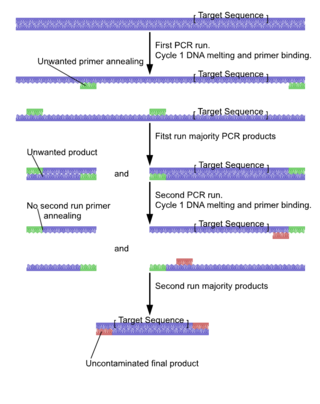
Nested polymerase chain reaction is a modification of polymerase chain reaction intended to reduce non-specific binding in products due to the amplification of unexpected primer binding sites.

Rolling circle replication (RCR) is a process of unidirectional nucleic acid replication that can rapidly synthesize multiple copies of circular molecules of DNA or RNA, such as plasmids, the genomes of bacteriophages, and the circular RNA genome of viroids. Some eukaryotic viruses also replicate their DNA or RNA via the rolling circle mechanism.
The selector technique is a method to amplify and multiplex genomic DNA.
The overlap extension polymerase chain reaction is a variant of PCR. It is also referred to as Splicing by overlap extension / Splicing by overhang extension (SOE) PCR. It is used assemble multiple smaller double stranded DNA fragments into a larger DNA sequence. OE-PCR is widely used to insert mutations at specific points in a sequence or to assemble custom DNA sequence from smaller DNA fragments into a larger polynucleotide.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
Multiplex ligation-dependent probe amplification (MLPA) is a variation of the multiplex polymerase chain reaction that permits amplification of multiple targets with only a single primer pair. It detects copy number changes at the molecular level, and software programs are used for analysis. Identification of deletions or duplications can indicate pathogenic mutations, thus MLPA is an important diagnostic tool used in clinical pathology laboratories worldwide.
A primer binding site is a region of a nucleotide sequence where an RNA or DNA single-stranded primer binds to start replication. The primer binding site is on one of the two complementary strands of a double-stranded nucleotide polymer, in the strand which is to be copied, or is within a single-stranded nucleotide polymer sequence.

The history of the polymerase chain reaction (PCR) has variously been described as a classic "Eureka!" moment, or as an example of cooperative teamwork between disparate researchers. Following is a list of events before, during, and after its development:
The versatility of polymerase chain reaction (PCR) has led to modifications of the basic protocol being used in a large number of variant techniques designed for various purposes. This article summarizes many of the most common variations currently or formerly used in molecular biology laboratories; familiarity with the fundamental premise by which PCR works and corresponding terms and concepts is necessary for understanding these variant techniques.
The ligase chain reaction (LCR) is a method of DNA amplification. The ligase chain reaction (LCR) is an amplification process that differs from PCR in that it involves a thermostable ligase to join two probes or other molecules together which can then be amplified by standard polymerase chain reaction (PCR) cycling. Each cycle results in a doubling of the target nucleic acid molecule. A key advantage of LCR is greater specificity as compared to PCR. Thus, LCR requires two completely different enzymes to operate properly: ligase, to join probe molecules together, and a thermostable polymerase to amplify those molecules involved in successful ligation. The probes involved in the ligation are designed such that the 5′ end of one probe is directly adjacent to the 3′ end of the other probe, thereby providing the requisite 3′-OH and 5′-PO4 group substrates for the ligase.
Diversity Arrays Technology (DArT) is a high-throughput genetic marker technique that can detect allelic variations to provides comprehensive genome coverage without any DNA sequence information for genotyping and other genetic analysis. The general steps involve reducing the complexity of the genomic DNA with specific restriction enzymes, choosing diverse fragments to serve as representations for the parent genomes, amplify via polymerase chain reaction (PCR), insert fragments into a vector to be placed as probes within a microarray, then fluorescent targets from a reference sequence will be allowed to hybridize with probes and put through an imaging system. The objective is to identify and quantify various forms of DNA polymorphism within genomic DNA of sampled species.

Vectorette PCR is a variation of polymerase chain reaction (PCR) designed in 1988. The original PCR was created and also patented during the 1980s. Vectorette PCR was first noted and described in an article in 1990 by John H. Riley and his team. Since then, multiple variants of PCR have been created. Vectorette PCR focuses on amplifying a specific sequence obtained from an internal sequence that is originally known until the fragment end. Multiple researches have taken this method as an opportunity to conduct experiments in order to uncover the potential uses that can be derived from Vectorette PCR.
