Related Research Articles

Creutzfeldt–Jakob disease (CJD), also known as subacute spongiform encephalopathy or neurocognitive disorder due to prion disease, is a fatal degenerative brain disorder. Early symptoms include memory problems, behavioral changes, poor coordination, and visual disturbances. Later symptoms include dementia, involuntary movements, blindness, weakness, and coma. About 70% of people die within a year of diagnosis. The name Creutzfeldt–Jakob disease was introduced by Walther Spielmeyer in 1922, after the German neurologists Hans Gerhard Creutzfeldt and Alfons Maria Jakob.

A prion is a misfolded protein that can transmit its misfoldedness to normal variants of the same protein and trigger cellular death. Prions cause prion diseases known as transmissible spongiform encephalopathies (TSEs) that are transmissible, fatal neurodegenerative diseases in humans and animals. The proteins may misfold sporadically, due to genetic mutations, or by exposure to an already misfolded protein. The consequent abnormal three-dimensional structure confers on them the ability to cause misfolding of other proteins.
Transmissible spongiform encephalopathies (TSEs) also known as prion diseases, are a group of progressive, incurable, and fatal conditions that are associated with prions and affect the brain and nervous system of many animals, including humans, cattle, and sheep. According to the most widespread hypothesis, they are transmitted by prions, though some other data suggest an involvement of a Spiroplasma infection. Mental and physical abilities deteriorate and many tiny holes appear in the cortex causing it to appear like a sponge when brain tissue obtained at autopsy is examined under a microscope. The disorders cause impairment of brain function, including memory changes, personality changes and problems with movement that worsen chronically.
Encephalopathy means any disorder or disease of the brain, especially chronic degenerative conditions. In modern usage, encephalopathy does not refer to a single disease, but rather to a syndrome of overall brain dysfunction; this syndrome has many possible organic and inorganic causes.
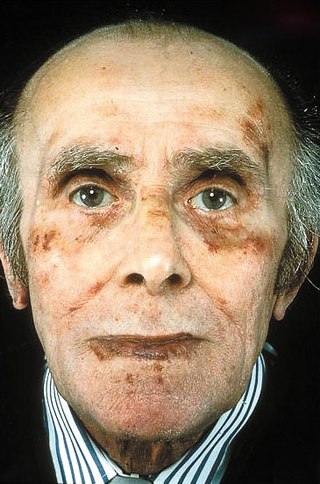
Amyloidosis is a group of diseases in which abnormal proteins, known as amyloid fibrils, build up in tissue. There are several non-specific and vague signs and symptoms associated with amyloidosis. These include fatigue, peripheral edema, weight loss, shortness of breath, palpitations, and feeling faint with standing. In AL amyloidosis, specific indicators can include enlargement of the tongue and periorbital purpura. In wild-type ATTR amyloidosis, non-cardiac symptoms include: bilateral carpal tunnel syndrome, lumbar spinal stenosis, biceps tendon rupture, small fiber neuropathy, and autonomic dysfunction.

Transthyretin (TTR or TBPA) is a transport protein in the plasma and cerebrospinal fluid that transports the thyroid hormone thyroxine (T4) and retinol to the liver. This is how transthyretin gained its name: transports thyroxine and retinol. The liver secretes TTR into the blood, and the choroid plexus secretes TTR into the cerebrospinal fluid.

Gerstmann–Sträussler–Scheinker syndrome (GSS) is an extremely rare, always fatal neurodegenerative disease that affects patients from 20 to 60 years in age. It is exclusively heritable, and is found in only a few families all over the world. It is, however, classified with the transmissible spongiform encephalopathies (TSE) due to the causative role played by PRNP, the human prion protein. GSS was first reported by the Austrian physicians Josef Gerstmann, Ernst Sträussler and Ilya Scheinker in 1936.

Amyloid plaques are extracellular deposits of the amyloid beta (Aβ) protein mainly in the grey matter of the brain. Degenerative neuronal elements and an abundance of microglia and astrocytes can be associated with amyloid plaques. Some plaques occur in the brain as a result of aging, but large numbers of plaques and neurofibrillary tangles are characteristic features of Alzheimer's disease. Abnormal neurites in amyloid plaques are tortuous, often swollen axons and dendrites. The neurites contain a variety of organelles and cellular debris, and many of them include characteristic paired helical filaments, the ultrastructural component of neurofibrillary tangles. The plaques are highly variable in shape and size; in tissue sections immunostained for Aβ, they comprise a log-normal size distribution curve with an average plaque area of 400-450 square micrometers (µm²). The smallest plaques, which often consist of diffuse deposits of Aβ, are particularly numerous. The apparent size of plaques is influenced by the type of stain used to detect them, and by the plane through which they are sectioned for analysis under the microscope. Plaques form when Aβ misfolds and aggregates into oligomers and longer polymers, the latter of which are characteristic of amyloid. Misfolded and aggregated Aβ is thought to be neurotoxic, especially in its oligomeric state.

Major prion protein(PrP) is encoded in the human body by the PRNP gene also known as CD230 (cluster of differentiation 230). Expression of the protein is most predominant in the nervous system but occurs in many other tissues throughout the body.

Tauopathy belongs to a class of neurodegenerative diseases involving the aggregation of tau protein into neurofibrillary or gliofibrillary tangles in the human brain. Tangles are formed by hyperphosphorylation of the microtubule protein known as tau, causing the protein to dissociate from microtubules and form insoluble aggregates. The mechanism of tangle formation is not well understood, and whether tangles are a primary cause of Alzheimer's disease or play a peripheral role is unknown.

Cardiac amyloidosis is a subcategory of amyloidosis where there is depositing of the protein amyloid in the cardiac muscle and surrounding tissues. Amyloid, a misfolded and insoluble protein, can become a deposit in the heart's atria, valves, or ventricles. These deposits can cause thickening of different sections of the heart, leading to decreased cardiac function. The overall decrease in cardiac function leads to a plethora of symptoms. This multisystem disease was often misdiagnosed, with a corrected analysis only during autopsy. Advancements of technologies have increased earlier accuracy of diagnosis. Cardiac amyloidosis has multiple sub-types including light chain, familial, and senile. One of the most studied types is light chain cardiac amyloidosis. Prognosis depends on the extent of the deposits in the body and the type of amyloidosis. New treatment methods are actively being researched in regards to the treatment of heart failure and specific cardiac amyloidosis problems.

In medicine, proteinopathy, or proteopathy, protein conformational disorder, or protein misfolding disease, is a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body. Often the proteins fail to fold into their normal configuration; in this misfolded state, the proteins can become toxic in some way or they can lose their normal function. The proteinopathies include such diseases as Creutzfeldt–Jakob disease and other prion diseases, Alzheimer's disease, Parkinson's disease, amyloidosis, multiple system atrophy, and a wide range of other disorders. The term proteopathy was first proposed in 2000 by Lary Walker and Harry LeVine.
Amyloid light-chain (AL) amyloidosis, also known as primary amyloidosis, is the most common form of systemic amyloidosis. The disease is caused when a person's antibody-producing cells do not function properly and produce abnormal protein fibers made of components of antibodies called light chains. These light chains come together to form amyloid deposits which can cause serious damage to different organs. An abnormal light chain in urine is known as Bence Jones protein.
AA amyloidosis is a form of amyloidosis, a disease characterized by the abnormal deposition of fibers of insoluble protein in the extracellular space of various tissues and organs. In AA amyloidosis, the deposited protein is serum amyloid A protein (SAA), an acute-phase protein which is normally soluble and whose plasma concentration is highest during inflammation.
The familial amyloid neuropathies are a rare group of autosomal dominant diseases wherein the autonomic nervous system and/or other nerves are compromised by protein aggregation and/or amyloid fibril formation.

Bovine spongiform encephalopathy (BSE), commonly known as mad cow disease, is an incurable and invariably fatal neurodegenerative disease of cattle. Symptoms include abnormal behavior, trouble walking, and weight loss. Later in the course of the disease the cow becomes unable to function normally. There is conflicting information about the time between infection and onset of symptoms. In 2002, the World Health Organization (WHO) suggested it to be approximately four to five years. Time from onset of symptoms to death is generally weeks to months. Spread to humans is believed to result in variant Creutzfeldt–Jakob disease (vCJD). As of 2018, a total of 231 cases of vCJD had been reported globally.
Familial amyloid cardiomyopathy (FAC), or transthyretin amyloid cardiomyopathy (ATTR-CM) results from the aggregation and deposition of mutant and wild-type transthyretin (TTR) protein in the heart. TTR is usually circulated as a homo-tetramer—a protein made up of four identical subunits—however, in FAC populations, TTR dissociates from this typical form and misassembles into amyloid fibrils which are insoluble and resistant to degradation. Due to this resistance to degradation, when amyloid fibrils accumulate in the heart's walls, specifically the left ventricle, rigidity prevents the heart from properly relaxing and refilling with blood: this is called diastolic dysfunction which can ultimately lead to heart failure.

LECT2 Amyloidosis (ALECT2) is a form of amyloidosis caused by the LECT2 protein. It was found to be the third most common cause of amyloidosis in a set of more than 4,000 individuals studied at the Mayo Clinic; the first and second most common forms the disorder were AL amyloidosis and AA amyloidosis, respectively. Amyloidosis is a disorder in which the abnormal deposition of a protein in organs and/or tissues gradually leads to organ failure and/or tissue injury.
Wild-type transthyretin amyloid (WTTA), also known as senile systemic amyloidosis (SSA), is a disease that typically affects the heart and tendons of elderly people. It is caused by the accumulation of a wild-type protein called transthyretin. This is in contrast to a related condition called transthyretin-related hereditary amyloidosis where a genetically mutated transthyretin protein tends to deposit much earlier than in WTTA due to abnormal conformation and bioprocessing. It belongs to a group of diseases called amyloidosis, chronic progressive conditions linked to abnormal deposition of normal or abnormal proteins, because these proteins are misshapen and cannot be properly degraded and eliminated by the cell metabolism.
Familial Danish Dementia is an extremely rare, neurodegenerative disease characterized by progressive cataracts, loss of hearing, cerebellar ataxia, paranoid psychosis, and dementia. Neuropathological hallmarks include extensive atrophy of all areas of the brain, chronic diffuse encephalopathy, and the presence of exceedingly thin and nearly totally demyelinated cranial nerves.