





Sphingomyelin phosphodiesterase 1 (SMPD1), also known as acid sphingomyelinase (ASM), is an enzyme that in humans is encoded by the SMPD1 gene.
Sphingomyelin phosphodiesterase 1 belongs to the sphingomyelin phosphodiesterase family. [5]
Sphingomyelin phosphodiesterase 1 (SMPD1), also known as acid sphingomyelinase (ASM), is an enzyme that in humans is encoded by the SMPD1 gene.
Sphingomyelin phosphodiesterase 1 belongs to the sphingomyelin phosphodiesterase family. [5]
Defects in the SMPD1 gene cause Niemann–Pick disease, SMPD1-associated. [5]
A mutation from leucine to proline at amino acid residue 302 encoded by the SMPD1 gene was identified by Gan-Or et al. (2013) as a risk factor for Parkinson disease. [6]

Tay–Sachs disease is a genetic disorder that results in the destruction of nerve cells in the brain and spinal cord. The most common form is infantile Tay–Sachs disease, which becomes apparent around the age of three to six months of age, with the baby losing the ability to turn over, sit, or crawl. This is then followed by seizures, hearing loss, and inability to move, with death usually occurring by the age of three to five. Less commonly, the disease may occur later in childhood, adolescence, or adulthood. These forms tend to be less severe, but the juvenile form typically results in death by age 15.

Niemann–Pick disease (NP), also known as acid sphingomyelinase deficiency, is a group of rare genetic diseases of varying severity. These are inherited metabolic disorders in which sphingomyelin accumulates in lysosomes in cells of many organs. NP types A, A/B, and B are cause by mutations in the SMPD1 gene, which causes a deficiency of a acid sphingomyelinase (ASM). NP type C is now considered a separate disease, as SMPD1 is not involved, and there is no deficiency in ASM.
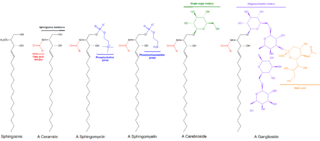
Sphingomyelin is a type of sphingolipid found in animal cell membranes, especially in the membranous myelin sheath that surrounds some nerve cell axons. It usually consists of phosphocholine and ceramide, or a phosphoethanolamine head group; therefore, sphingomyelins can also be classified as sphingophospholipids. In humans, SPH represents ~85% of all sphingolipids, and typically make up 10–20 mol % of plasma membrane lipids.

α-Galactosidase is a glycoside hydrolase enzyme that catalyses the following reaction:

Sphingolipidoses are a class of lipid storage disorders or degenerative storage disorders caused by deficiency of an enzyme that is required for the catabolism of lipids that contain ceramide, also relating to sphingolipid metabolism. The main members of this group are Niemann–Pick disease, Fabry disease, Krabbe disease, Gaucher disease, Tay–Sachs disease and metachromatic leukodystrophy. They are generally inherited in an autosomal recessive fashion, but notably Fabry disease is X-linked recessive. Taken together, sphingolipidoses have an incidence of approximately 1 in 10,000, but substantially more in certain populations such as Ashkenazi Jews. Enzyme replacement therapy is available to treat mainly Fabry disease and Gaucher disease, and people with these types of sphingolipidoses may live well into adulthood. The other types are generally fatal by age 1 to 5 years for infantile forms, but progression may be mild for juvenile- or adult-onset forms.

Sphingomyelin phosphodiesterase is a hydrolase enzyme that is involved in sphingolipid metabolism reactions. SMase is a member of the DNase I superfamily of enzymes and is responsible for breaking sphingomyelin (SM) down into phosphocholine and ceramide. The activation of SMase has been suggested as a major route for the production of ceramide in response to cellular stresses.
The GM2 gangliosidoses are a group of three related genetic disorders that result from a deficiency of the enzyme beta-hexosaminidase. This enzyme catalyzes the biodegradation of fatty acid derivatives known as gangliosides. The diseases are better known by their individual names: Tay–Sachs disease, AB variant, and Sandhoff disease.

Hexosaminidase is an enzyme involved in the hydrolysis of terminal N-acetyl-D-hexosamine residues in N-acetyl-β-D-hexosaminides.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.

Niemann-Pick disease, type C1 (NPC1) is a membrane protein that mediates intracellular cholesterol trafficking in mammals. In humans the protein is encoded by the NPC1 gene.

Hexosaminidase A (alpha polypeptide), also known as HEXA, is an enzyme that in humans is encoded by the HEXA gene, located on the 15th chromosome.

The ASAH1 gene encodes in humans the acid ceramidase enzyme.

Ectonucleotide pyrophosphatase/phosphodiesterase family member 7 also known as alkaline sphingomyelin phosphodiesterase (Alk-SMase) or intestinal alkaline sphingomyelinase is an enzyme that in humans is encoded by the ENPP7 gene.

Sphingomyelin phosphodiesterase 3 is an enzyme that in humans is encoded by the SMPD3 gene.

Sphingomyelin phosphodiesterase 2 is an enzyme that in humans is encoded by the SMPD2 gene.
Niemann–Pick type C (NPC) is a lysosomal storage disease associated with mutations in NPC1 and NPC2 genes. Niemann–Pick type C affects an estimated 1:150,000 people. Approximately 50% of cases present before 10 years of age, but manifestations may first be recognized as late as the sixth decade.
Niemann–Pick disease, SMPD1-associated refers to two different types of Niemann–Pick disease, type A (NPA) and type B (NPB), which are associated with the SMPD1 gene.

Robert J. Desnick is an American human geneticist whose basic and translational research accomplishments include significant discoveries in genomics, pharmacogenetics, gene therapy, personalized medicine, and the treatment of genetic diseases. His translational research has led to the development of the enzyme replacement therapy (ERT) and the chaperone therapy for Fabry disease, ERT for Niemann–Pick disease type B, and the RNA Interference Therapy for the Acute Hepatic Porphyrias.
Functional inhibitors of acid sphingomyelinase, or FIASMA, is a large group of pharmacological compounds inhibiting the enzyme acid sphingomyelinase. This enzyme is mainly located within the lysosome, where it cleaves sphingomyelin to ceramide and sphingosine, the latter of which is then phosphorylated to sphingosine-1-phosphate. These metabolites, and subsequent inhibition of the enzyme, influence the balance between cell death (apoptosis) and cell growth (proliferation). A lack of regulation of this sensitive equilibrium can lead to serious clinical consequences.
Acid sphingomyelinase is one of the enzymes that make up the sphingomyelinase (SMase) family, responsible for catalyzing the breakdown of sphingomyelin to ceramide and phosphorylcholine. They are organized into alkaline, neutral, and acidic SMase depending on the pH in which their enzymatic activity is optimal. Acid sphingomyelinases' (aSMases) enzymatic activity can be influenced by drugs, lipids, cations, pH, redox and other proteins in the environment. Specifically aSMases have been shown to have increased enzymatic activity in lysobisphosphatidic acid (LBPA) or phosphatidylinositol (PI) enriched environments, and inhibited activity when phosphorylated derivatives of PI are present.
| | This article on a gene on human chromosome 11 is a stub. You can help Wikipedia by expanding it. |