Signs and symptoms
| | This section is empty. You can help by adding to it. (November 2021) |
| Sticky platelet syndrome | |
|---|---|
| Specialty | Hematology |
Sticky platelet syndrome is a term used by some [1] [2] [3] [4] to describe a disorder of platelet function. [5] It was first described by Mammen in 1983. [6] It is inherited in an autosomal dominant pattern. [7] It has not been associated with a specific gene, and it is not recognized as an entity in OMIM.
Among researchers using the term, it has been described as a coagulation disorder that can present in conjunction with protein S deficiency and Factor V Leiden. [8] It is not currently known if sticky platelet syndrome is a distinct condition, or if it represents part of the presentation of a more well characterized coagulation disorder.
| | This section is empty. You can help by adding to it. (November 2021) |
The syndrome is believed to be hereditary. [9]
SPS is diagnosed by demonstrating platelet hyperaggregability. In a lab test called aggregometry platelet stickiness is stimulated with epinephrine (EPI) and/or adenosine diphosphate (ADP). [10] This test is not possible for patients being treated with acetylsalicylic acid until that substance has sufficiently cleared from their system.[ citation needed ]
Those diagnosed are usually treated with taking a low dose (80–100 mg) Aspirin a day. [11] Anticoagulants (e.g. Warfarin, Coumadin) or clopidogrel (Plavix) are often additionally prescribed following formation of a medically significant clot. Thrombelastography is more commonly being used to diagnose hypercoagulability and monitor anti-platelet therapy.[ citation needed ]

Coagulation, also known as clotting, is the process by which blood changes from a liquid to a gel, forming a blood clot. It potentially results in hemostasis, the cessation of blood loss from a damaged vessel, followed by repair. The mechanism of coagulation involves activation, adhesion and aggregation of platelets, as well as deposition and maturation of fibrin.

Polycythemia vera is an uncommon myeloproliferative neoplasm in which the bone marrow makes too many red blood cells. It may also result in the overproduction of white blood cells and platelets.
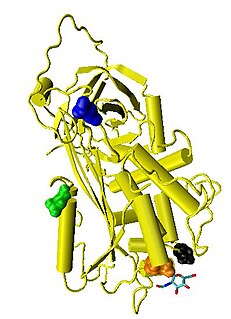
Antithrombin (AT) is a small glycoprotein that inactivates several enzymes of the coagulation system. It is a 432-amino-acid protein produced by the liver. It contains three disulfide bonds and a total of four possible glycosylation sites. α-Antithrombin is the dominant form of antithrombin found in blood plasma and has an oligosaccharide occupying each of its four glycosylation sites. A single glycosylation site remains consistently un-occupied in the minor form of antithrombin, β-antithrombin. Its activity is increased manyfold by the anticoagulant drug heparin, which enhances the binding of antithrombin to factor IIa (Thrombin) and factor Xa.

In medicine (hematology), bleeding diathesis is an unusual susceptibility to bleed (hemorrhage) mostly due to hypocoagulability, in turn caused by a coagulopathy. Therefore, this may result in the reduction of platelets being produced and leads to excessive bleeding. Several types of coagulopathy are distinguished, ranging from mild to lethal. Coagulopathy can be caused by thinning of the skin, such that the skin is weakened and is bruised easily and frequently without any trauma or injury to the body. Also, coagulopathy can be contributed by impaired wound healing or impaired clot formation.

Protein S is a vitamin K-dependent plasma glycoprotein synthesized in the liver. In the circulation, Protein S exists in two forms: a free form and a complex form bound to complement protein C4b-binding protein (C4BP). In humans, protein S is encoded by the PROS1 gene. Protein S plays a role in coagulation.
Thrombophilia is an abnormality of blood coagulation that increases the risk of thrombosis. Such abnormalities can be identified in 50% of people who have an episode of thrombosis that was not provoked by other causes. A significant proportion of the population has a detectable thrombophilic abnormality, but most of these develop thrombosis only in the presence of an additional risk factor.
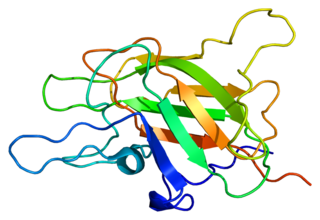
Factor V is a protein of the coagulation system, rarely referred to as proaccelerin or labile factor. In contrast to most other coagulation factors, it is not enzymatically active but functions as a cofactor. Deficiency leads to predisposition for hemorrhage, while some mutations predispose for thrombosis.

Factor XI or plasma thromboplastin antecedent is the zymogen form of factor XIa, one of the enzymes of the coagulation cascade. Like many other coagulation factors, it is a serine protease. In humans, Factor XI is encoded by the F11 gene.

Activated protein C resistance (APCR) is a hypercoagulability characterized by a lack of a response to activated protein C (APC), which normally helps prevent blood from clotting excessively. This results in an increased risk of venous thrombosis, which resulting in medical conditions such as deep vein thrombosis and pulmonary embolism.

Protein C deficiency is a rare genetic trait that predisposes to thrombotic disease. It was first described in 1981. The disease belongs to a group of genetic disorders known as thrombophilias. Protein C deficiency is associated with an increased incidence of venous thromboembolism, whereas no association with arterial thrombotic disease has been found.

Factor X deficiency is a bleeding disorder characterized by a lack in the production of factor X (FX), an enzyme protein that causes blood to clot in the coagulation cascade. Produced in the liver FX when activated cleaves prothrombin to generate thrombin in the intrinsic pathway of coagulation. This process is vitamin K dependent and enhanced by activated factor V.
Factor VII deficiency is a bleeding disorder characterized by a lack in the production of Factor VII (FVII) (proconvertin), a protein that causes blood to clot in the coagulation cascade. After a trauma factor VII initiates the process of coagulation in conjunction with tissue factor in the extrinsic pathway.
Purpura fulminans is an acute, often fatal, thrombotic disorder which manifests as blood spots, bruising and discolouration of the skin resulting from coagulation in small blood vessels within the skin and rapidly leads to skin necrosis and disseminated intravascular coagulation.

β2-glycoprotein 1, also known as beta-2 glycoprotein 1 and Apolipoprotein H (Apo-H), is a 38 kDa multifunctional plasma protein that in humans is encoded by the APOH gene. One of its functions is to bind cardiolipin. When bound, the structure of cardiolipin and β2-GP1 both undergo large changes in structure. Within the structure of Apo-H is a stretch of positively charged amino acids, Lys-Asn-Lys-Glu-Lys-Lys, are involved in phospholipid binding.
The dysfibrinogenemias consist of three types of fibrinogen disorders in which a critical blood clotting factor, fibrinogen, circulates at normal levels but is dysfunctional. Congenital dysfibrinogenemia is an inherited disorder in which one of the parental genes produces an abnormal fibrinogen. This fibrinogen interferes with normal blood clotting and/or lysis of blood clots. The condition therefore may cause pathological bleeding and/or thrombosis. Acquired dysfibrinogenemia is a non-hereditary disorder in which fibrinogen is dysfunctional due to the presence of liver disease, autoimmune disease, a plasma cell dyscrasias, or certain cancers. It is associated primarily with pathological bleeding. Hereditary fibrinogen Aα-Chain amyloidosis is a sub-category of congenital dysfibrinogenemia in which the dysfunctional fibrinogen does not cause bleeding or thrombosis but rather gradually accumulates in, and disrupts the function of, the kidney.

Coagulation factor XIII A chain is a protein that in humans is encoded by the F13A1 gene.
Atypical hemolytic uremic syndrome (aHUS) is an extremely rare, life-threatening, progressive disease that frequently has a genetic component. In most cases it can be effectively controlled by interruption of the complement cascade. Particular monoclonal antibodies, discussed later in the article, have proven efficacy in many cases.
Blood clotting tests are the tests used for diagnostics of the hemostasis system. Coagulometer is the medical laboratory analyzer used for testing of the hemostasis system. Modern coagulometers realize different methods of activation and observation of development of blood clots in blood or in blood plasma.

Upshaw–Schulman syndrome (USS) is the recessively inherited form of thrombotic thrombocytopenic purpura (TTP), a rare and complex blood coagulation disease. USS is caused by the absence of the ADAMTS13 protease resulting in the persistence of ultra large von Willebrand factor multimers (ULVWF), causing episodes of acute thrombotic microangiopathy with disseminated multiple small vessel obstructions. These obstructions deprive downstream tissues from blood and oxygen, which can result in tissue damage and death. The presentation of an acute USS episode is variable but usually associated with thrombocytopenia, microangiopathic hemolytic anemia (MAHA) with schistocytes on the peripheral blood smear, fever and signs of ischemic organ damage in the brain, kidney and heart.

Björn Dahlbäck is a Swedish physician, medical researcher, and professor of clinical chemistry, specializing in hematology and the molecular mechanisms of blood coagulation. He is perhaps "best known for his groundbreaking discovery of activated protein C (APC) resistance as the most common inherited risk factor of venous thrombosis."