
The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.

Genomics is an interdisciplinary field of biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.
The International HapMap Project was an organization that aimed to develop a haplotype map (HapMap) of the human genome, to describe the common patterns of human genetic variation. HapMap is used to find genetic variants affecting health, disease and responses to drugs and environmental factors. The information produced by the project is made freely available for research.
Indel (insertion-deletion) is a molecular biology term for an insertion or deletion of bases in the genome of an organism. Indels ≥ 50 bases in length are classified as structural variants.

Human genetic variation is the genetic differences in and among populations. There may be multiple variants of any given gene in the human population (alleles), a situation called polymorphism.
Human evolutionary genetics studies how one human genome differs from another human genome, the evolutionary past that gave rise to the human genome, and its current effects. Differences between genomes have anthropological, medical, historical and forensic implications and applications. Genetic data can provide important insights into human evolution.

In genomics, a genome-wide association study, is an observational study of a genome-wide set of genetic variants in different individuals to see if any variant is associated with a trait. GWA studies typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like major human diseases, but can equally be applied to any other genetic variants and any other organisms.
Minor allele frequency (MAF) is the frequency at which the second most common allele occurs in a given population. They play a surprising role in heritability since MAF variants which occur only once, known as "singletons", drive an enormous amount of selection.
Population genomics is the large-scale comparison of DNA sequences of populations. Population genomics is a neologism that is associated with population genetics. Population genomics studies genome-wide effects to improve our understanding of microevolution so that we may learn the phylogenetic history and demography of a population.
Whole genome sequencing (WGS), also known as full genome sequencing, complete genome sequencing, or entire genome sequencing, is the process of determining the entirety, or nearly the entirety, of the DNA sequence of an organism's genome at a single time. This entails sequencing all of an organism's chromosomal DNA as well as DNA contained in the mitochondria and, for plants, in the chloroplast.

Exome sequencing, also known as whole exome sequencing (WES), is a genomic technique for sequencing all of the protein-coding regions of genes in a genome. It consists of two steps: the first step is to select only the subset of DNA that encodes proteins. These regions are known as exons—humans have about 180,000 exons, constituting about 1% of the human genome, or approximately 30 million base pairs. The second step is to sequence the exonic DNA using any high-throughput DNA sequencing technology.

A reference genome is a digital nucleic acid sequence database, assembled by scientists as a representative example of the set of genes in one idealized individual organism of a species. As they are assembled from the sequencing of DNA from a number of individual donors, reference genomes do not accurately represent the set of genes of any single individual organism. Instead, a reference provides a haploid mosaic of different DNA sequences from each donor. For example, one of the most recent human reference genomes, assembly GRCh38/hg38, is derived from >60 genomic clone libraries. There are reference genomes for multiple species of viruses, bacteria, fungus, plants, and animals. Reference genomes are typically used as a guide on which new genomes are built, enabling them to be assembled much more quickly and cheaply than the initial Human Genome Project. Reference genomes can be accessed online at several locations, using dedicated browsers such as Ensembl or UCSC Genome Browser.
Genomic structural variation is the variation in structure of an organism's chromosome. It consists of many kinds of variation in the genome of one species, and usually includes microscopic and submicroscopic types, such as deletions, duplications, copy-number variants, insertions, inversions and translocations. Originally, a structure variation affects a sequence length about 1kb to 3Mb, which is larger than SNPs and smaller than chromosome abnormality. However, the operational range of structural variants has widened to include events > 50bp. The definition of structural variation does not imply anything about frequency or phenotypical effects. Many structural variants are associated with genetic diseases, however many are not. Recent research about SVs indicates that SVs are more difficult to detect than SNPs. Approximately 13% of the human genome is defined as structurally variant in the normal population, and there are at least 240 genes that exist as homozygous deletion polymorphisms in human populations, suggesting these genes are dispensable in humans. Rapidly accumulating evidence indicates that structural variations can comprise millions of nucleotides of heterogeneity within every genome, and are likely to make an important contribution to human diversity and disease susceptibility.

Jumping libraries or junction-fragment libraries are collections of genomic DNA fragments generated by chromosome jumping. These libraries allow the analysis of large areas of the genome and overcome distance limitations in common cloning techniques. A jumping library clone is composed of two stretches of DNA that are usually located many kilobases away from each other. The stretch of DNA located between these two "ends" is deleted by a series of biochemical manipulations carried out at the start of this cloning technique.
Philip Awadalla is a professor of medical and population genetics at the Ontario Institute for Cancer Research, and the Department of Molecular Genetics, Faculty of Medicine, University of Toronto. He is the National Scientific Director of the Canadian Partnership for Tomorrow's Health (CanPath), formerly the Canadian Partnership for Tomorrow Project (CPTP), and executive director of the Ontario Health Study. He is also the Executive Scientific Director of the Genome Canada Genome Technology Platform, the Canadian Data Integration Centre. Professor Awadalla was the Executive Scientific Director of the CARTaGENE biobank, a regional cohort member of the CPTP, from 2009 to 2015, and is currently a scientific advisor for this and other scientific and industry platforms. At the OICR, he is Director of Computational Biology.
Imputation in genetics refers to the statistical inference of unobserved genotypes. It is achieved by using known haplotypes in a population, for instance from the HapMap or the 1000 Genomes Project in humans, thereby allowing to test for association between a trait of interest and experimentally untyped genetic variants, but whose genotypes have been statistically inferred ("imputed"). Genotype imputation is usually performed on SNPs, the most common kind of genetic variation.
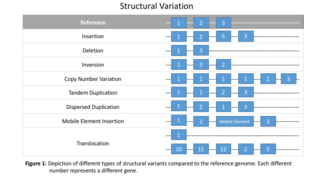
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic. This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.
ANNOVAR is a bioinformatics software tool for the interpretation and prioritization of single nucleotide variants (SNVs), insertions, deletions, and copy number variants (CNVs) of a given genome.


