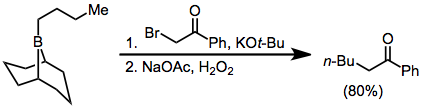
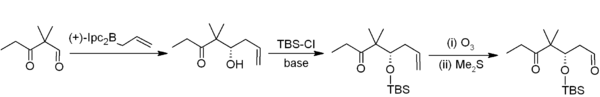Hydroboration–oxidation reaction is a two-step hydration reaction that converts an alkene into an alcohol. The process results in the syn addition of a hydrogen and a hydroxyl group where the double bond had been. Hydroboration–oxidation is an anti-Markovnikov reaction, with the hydroxyl group attaching to the less-substituted carbon. The reaction thus provides a more stereospecific and complementary regiochemical alternative to other hydration reactions such as acid-catalyzed addition and the oxymercuration–reduction process. The reaction was first reported by Herbert C. Brown in the late 1950s and it was recognized in his receiving the Nobel Prize in Chemistry in 1979.
The Suzuki reaction is an organic reaction, classified as a cross-coupling reaction, where the coupling partners are a boronic acid and an organohalide and the catalyst is a palladium(0) complex. It was first published in 1979 by Akira Suzuki, and he shared the 2010 Nobel Prize in Chemistry with Richard F. Heck and Ei-ichi Negishi for their contribution to the discovery and development of palladium-catalyzed cross-couplings in organic synthesis. This reaction is also known as the Suzuki–Miyaura reaction or simply as the Suzuki coupling. It is widely used to synthesize polyolefins, styrenes, and substituted biphenyls. Several reviews have been published describing advancements and the development of the Suzuki reaction. The general scheme for the Suzuki reaction is shown below, where a carbon-carbon single bond is formed by coupling a halide (R1-X) with an organoboron species (R2-BY2) using a palladium catalyst and a base. The organoboron species is usually synthesized by hydroboration or carboboration, allowing for rapid generation of molecular complexity.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
In organic chemistry, hydroboration refers to the addition of a hydrogen-boron bond to certain double and triple bonds involving carbon. This chemical reaction is useful in the organic synthesis of organic compounds.

Nucleophilic conjugate addition is a type of organic reaction. Ordinary nucleophilic additions or 1,2-nucleophilic additions deal mostly with additions to carbonyl compounds. Simple alkene compounds do not show 1,2 reactivity due to lack of polarity, unless the alkene is activated with special substituents. With α,β-unsaturated carbonyl compounds such as cyclohexenone it can be deduced from resonance structures that the β position is an electrophilic site which can react with a nucleophile. The negative charge in these structures is stored as an alkoxide anion. Such a nucleophilic addition is called a nucleophilic conjugate addition or 1,4-nucleophilic addition. The most important active alkenes are the aforementioned conjugated carbonyls and acrylonitriles.

The Petasis reaction is the multi-component reaction of an amine, a carbonyl, and a vinyl- or aryl-boronic acid to form substituted amines.

A boronic acid is an organic compound related to boric acid in which one of the three hydroxyl groups is replaced by an alkyl or aryl group. As a compound containing a carbon–boron bond, members of this class thus belong to the larger class of organoboranes.

Phenylboronic acid or benzeneboronic acid, abbreviated as PhB(OH)2 where Ph is the phenyl group C6H5-, is a boronic acid containing a phenyl substituent and two hydroxyl groups attached to boron. Phenylboronic acid is a white powder and is commonly used in organic synthesis. Boronic acids are mild Lewis acids which are generally stable and easy to handle, making them important to organic synthesis.
In organometallic chemistry, a migratory insertion is a type of reaction wherein two ligands on a metal complex combine. It is a subset of reactions that very closely resembles the insertion reactions, and both are differentiated by the mechanism that leads to the resulting stereochemistry of the products. However, often the two are used interchangeably because the mechanism is sometimes unknown. Therefore, migratory insertion reactions or insertion reactions, for short, are defined not by the mechanism but by the overall regiochemistry wherein one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:

Diisopinocampheylborane is an organoborane that is useful for asymmetric synthesis. This colourless solid is the precursor to a range of related reagents. The compound was reported in 1961 by Zweifel and Brown in a pioneering demonstration of asymmetric synthesis using boranes. The reagent is mainly used for the synthesis of chiral secondary alcohols. The reagent is often depicted as a monomer but like most hydroboranes, it is dimeric with B-H-B bridges.

Alpine borane is the commercial name for an organoboron compound that is used in organic synthesis. It is a colorless liquid, although it is usually encountered as a solution. A range of alkyl-substituted borane are specialty reagents in organic synthesis. Two such reagents that are closely related to Alpine borane are 9-BBN and diisopinocampheylborane.

The Liebeskind–Srogl coupling reaction is an organic reaction forming a new carbon–carbon bond from a thioester and a boronic acid using a metal catalyst. It is a cross-coupling reaction. This reaction was invented by and named after Jiri Srogl from the Academy of Sciences, Czech Republic, and Lanny S. Liebeskind from Emory University, Atlanta, Georgia, USA. There are three generations of this reaction, with the first generation shown below. The original transformation used catalytic Pd(0), TFP = tris(2-furyl)phosphine as an additional ligand and stoichiometric CuTC = copper(I) thiophene-2-carboxylate as a co-metal catalyst. The overall reaction scheme is shown below.

Borane dimethylsulfide (BMS) is a chemical compound with the chemical formula BH3·S(CH3)2. It is an adduct between borane molecule and dimethyl sulfide molecule. It is a complexed borane reagent that is used for hydroborations and reductions. The advantages of BMS over other borane reagents, such as borane-tetrahydrofuran, are its increased stability and higher solubility. BMS is commercially available at much higher concentrations than its tetrahydrofuran counterpart and does not require sodium borohydride as a stabilizer, which could result in undesired side reactions. In contrast, BH3·THF requires sodium borohydride to inhibit reduction of THF to tributyl borate. BMS is soluble in most aprotic solvents.
Electrophilic amination is a chemical process involving the formation of a carbon–nitrogen bond through the reaction of a nucleophilic carbanion with an electrophilic source of nitrogen.
Reactions of alkenyl- and alkynylaluminium compounds involve the transfer of a nucleophilic alkenyl or alkynyl group attached to aluminium to an electrophilic atom. Stereospecific hydroalumination, carboalumination, and terminal alkyne metalation are useful methods for generation of the necessary alkenyl- and alkynylalanes.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.

Protodeboronation, or protodeborylation is a chemical reaction involving the protonolysis of a boronic acid in which a carbon-boron bond is broken and replaced with a carbon-hydrogen bond. Protodeboronation is a well-known undesired side reaction, and frequently associated with metal-catalysed coupling reactions that utilise boronic acids. For a given boronic acid, the propensity to undergo protodeboronation is highly variable and dependent on various factors, such as the reaction conditions employed and the organic substituent of the boronic acid.
Norio Miyaura was a Japanese organic chemist. He was a professor of graduate chemical engineering at Hokkaido University. His major accomplishments surrounded his work in cross-coupling reactions / conjugate addition reactions of organoboronic acids and addition / coupling reactions of diborons and boranes. He is also the co-author of Cross-Coupling Reactions: A Practical Guide with M. Nomura E. S.. Miyaura was a world-known and accomplished researcher by the time he retired and so, in 2007, he won the Japan Chemical Society Award.
In organic chemistry, carboboration describes an addition of both a carbon and a boron moiety to certain carbon-containing double and triple bonds, such as alkenes, alkynes, and allenes.