Related Research Articles
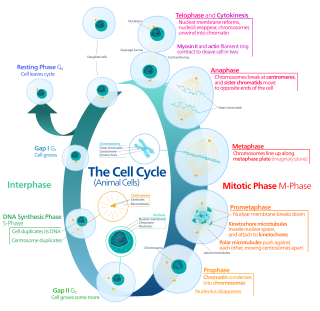
The G0 phase describes a cellular state outside of the replicative cell cycle. Classically, cells were thought to enter G0 primarily due to environmental factors, like nutrient deprivation, that limited the resources necessary for proliferation. Thus it was thought of as a resting phase. G0 is now known to take different forms and occur for multiple reasons. For example, most adult neuronal cells, among the most metabolically active cells in the body, are fully differentiated and reside in a terminal G0 phase. Neurons reside in this state, not because of stochastic or limited nutrient supply, but as a part of their developmental program.

The mammalian target of rapamycin (mTOR), also referred to as the mechanistic target of rapamycin, and sometimes called FK506-binding protein 12-rapamycin-associated protein 1 (FRAP1), is a kinase that in humans is encoded by the MTOR gene. mTOR is a member of the phosphatidylinositol 3-kinase-related kinase family of protein kinases.

Interleukin-1 alpha also known as hematopoietin 1 is a cytokine of the interleukin 1 family that in humans is encoded by the IL1A gene. In general, Interleukin 1 is responsible for the production of inflammation, as well as the promotion of fever and sepsis. IL-1α inhibitors are being developed to interrupt those processes and treat diseases.

p16, is a protein that slows cell division by slowing the progression of the cell cycle from the G1 phase to the S phase, thereby acting as a tumor suppressor. It is encoded by the CDKN2A gene. A deletion in this gene can result in insufficient or non-functional p16, accelerating the cell cycle and resulting in many types of cancer.
Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription machinery proteins, and thereby control gene expression. Such remodeling is principally carried out by 1) covalent histone modifications by specific enzymes, e.g., histone acetyltransferases (HATs), deacetylases, methyltransferases, and kinases, and 2) ATP-dependent chromatin remodeling complexes which either move, eject or restructure nucleosomes. Besides actively regulating gene expression, dynamic remodeling of chromatin imparts an epigenetic regulatory role in several key biological processes, egg cells DNA replication and repair; apoptosis; chromosome segregation as well as development and pluripotency. Aberrations in chromatin remodeling proteins are found to be associated with human diseases, including cancer. Targeting chromatin remodeling pathways is currently evolving as a major therapeutic strategy in the treatment of several cancers.

An aging-associated disease is a disease that is most often seen with increasing frequency with increasing senescence. They are essentially complications of senescence, distinguished from the aging process itself because all adult animals age but not all adult animals experience all age-associated diseases. The term does not refer to age-specific diseases, such as the childhood diseases chicken pox and measles, only diseases of the elderly. They are also not accelerated aging diseases, all of which are genetic disorders.
Caretaker genes encode products that stabilize the genome. Fundamentally, mutations in caretaker genes lead to genomic instability. Tumor cells arise from two distinct classes of genomic instability: mutational instability arising from changes in the nucleotide sequence of DNA and chromosomal instability arising from improper rearrangement of chromosomes.

MAP kinase-activated protein kinase 2 is an enzyme that in humans is encoded by the MAPKAPK2 gene.

High-mobility group protein B2 also known as high-mobility group protein 2 (HMG-2) is a protein that in humans is encoded by the HMGB2 gene.
Butyrate response factor 1 is a protein that in humans is encoded by the ZFP36L1 gene.
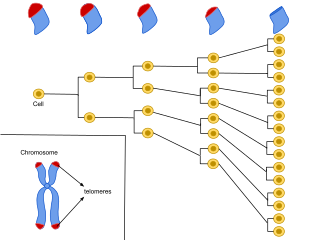
Cellular senescence is a phenomenon characterized by the cessation of cell division. In their experiments during the early 1960s, Leonard Hayflick and Paul Moorhead found that normal human fetal fibroblasts in culture reach a maximum of approximately 50 cell population doublings before becoming senescent. This process is known as "replicative senescence", or the Hayflick limit. Hayflick's discovery of mortal cells paved the path for the discovery and understanding of cellular aging molecular pathways. Cellular senescence can be initiated by a wide variety of stress inducing factors. These stress factors include both environmental and internal damaging events, abnormal cellular growth, oxidative stress, autophagy factors, among many other things.

Yippee-like 3 (Drosophila) is a protein that in humans is encoded by the YPEL3 gene. YPEL3 has growth inhibitory effects in normal and tumor cell lines. One of five family members (YPEL1-5), YPEL3 was named in reference to its Drosophila melanogaster orthologue. Initially discovered in a gene expression profiling assay of p53 activated MCF7 cells, induction of YPEL3 has been shown to trigger permanent growth arrest or cellular senescence in certain human normal and tumor cell types. DNA methylation of a CpG island near the YPEL3 promoter as well as histone acetylation may represent possible epigenetic mechanisms leading to decreased gene expression in human tumors.
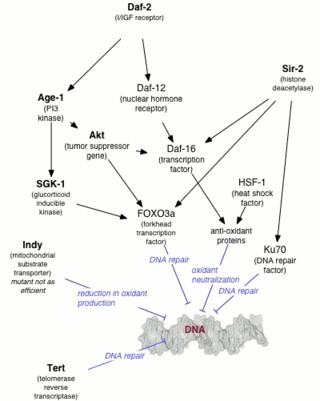
Genetics of aging is generally concerned with life extension associated with genetic alterations, rather than with accelerated aging diseases leading to reduction in lifespan.
Androgen deprivation-induced senescence refers to the induction of cellular senescence as a result of androgen deprivation therapy. ADIS is observed in prostate cancer cells that are dependent on androgens for cell proliferation. Androgen withdrawal induces cells to undergo cellular senescence by up-regulating intracellular reactive oxygen species (ROS) that cause DNA damage. ADIS is maintained through the up-regulation of the cell cycle regulator p16ink4a.
An epigenetic clock is a biochemical test that can be used to measure age. The test is based on DNA methylation levels, measuring the accumulation of methyl groups to one's DNA molecules.
A senolytic is among a class of small molecules under basic research to determine if they can selectively induce death of senescent cells and improve health in humans. A goal of this research is to discover or develop agents to delay, prevent, alleviate, or reverse age-related diseases. Removal of senescent cells with senolytics has been proposed as a method of enhancing immunity during aging.
Judith Campisi was an American biochemist and cell biologist. She was a professor of biogerontology at the Buck Institute for Research on Aging. She was also a member of the SENS Research Foundation Advisory Board and an adviser at the Lifeboat Foundation. She was co-editor in chief of the Aging Journal, together with Mikhail Blagosklonny and David Sinclair, and founder of the pharmaceutical company Unity Biotechnology. She is listed in Who's Who in Gerontology. She was widely known for her research on how senescent cells influence aging and cancer — in particular the Senescence Associated Secretory Phenotype (SASP).
Senotherapeutic's refers to therapeutic agents/strategies that specifically target cellular senescence. Senotherapeutic's include emerging senolytic/senoptotic small molecules that specifically induce cell death in senescent cells and agents that inhibit the pro-inflammatory senescent secretome. Senescent cells can be targeted for immune clearance, but an ageing immune system likely impairs senescent cell clearance leading to their accumulation. Therefore, agents which can enhance immune clearance of senescent cells can also be considered as senotherapeutic.
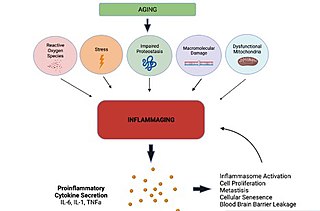
Inflammaging is a chronic, sterile, low-grade inflammation that develops with advanced age, in the absence of overt infection, and may contribute to clinical manifestations of other age-related pathologies. Inflammaging is thought to be caused by a loss of control over systemic inflammation resulting in chronic overstimulation of the innate immune system. Inflammaging is a significant risk factor in mortality and morbidity in aged individuals.
Aging is characterized by a progressive loss of physiological integrity, leading to impaired function and increased vulnerability to death. The hallmarks of aging are the types of biochemical changes that occur in all organisms that experience biological aging and lead to a progressive loss of physiological integrity, impaired function and, eventually, death. They were first listed in a landmark paper in 2013 to conceptualize the essence of biological aging and its underlying mechanisms.
References
- 1 2 3 Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008). "Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor". PLOS Biology . 6 (12): 2853–2868. doi: 10.1371/journal.pbio.0060301 . PMC 2592359 . PMID 19053174.
- 1 2 3 Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM (2017). "Senescent cells: an emerging target for diseases of ageing". Nature Reviews Drug Discovery . 16 (10): 718–735. doi:10.1038/nrd.2017.116. PMC 5942225 . PMID 28729727.
- 1 2 Prata LG, Ovsyannikova IG, Tchkonia T, Kirkland JL (2018). "Senescent cell clearance by the immune system: Emerging therapeutic opportunities". Seminars in Immunology . 40: 101275. doi:10.1016/j.smim.2019.04.003. PMC 7061456 . PMID 31088710.
- 1 2 3 Birch J, Gil J (2020). "Senescence and the SASP: many therapeutic avenues". Genes & Development . 34 (23–24): 1565–1576. doi:10.1101/gad.343129.120. PMC 7706700 . PMID 33262144.
- ↑ Amor C, Feucht J, Lowe SW (2020). "Senolytic CAR T cells reverse senescence-associated pathologies". Nature . 583 (7814): 127–132. Bibcode:2020Natur.583..127A. doi:10.1038/s41586-020-2403-9. PMC 7583560 . PMID 32555459.
- ↑ Ito Y, Hoare M, Narita M (2017). "Spatial and Temporal Control of Senescence". Trends in Cell Biology . 27 (11): 820–832. doi:10.1016/j.tcb.2017.07.004. PMID 28822679.
- 1 2 Nacarelli T, Lau L, Fukumoto T, David G, Zhang R (2019). "NAD + metabolism governs the proinflammatory senescence-associated secretome". Nature Cell Biology . 21 (3): 397–407. doi:10.1038/s41556-019-0287-4. PMC 6448588 . PMID 30778219.
- 1 2 Basisty N, Kale A, Jeon O, Kuehnemann C, Payne T, Rao C, Holtz A, Shah S, Vagisha Sharma V, Ferrucci L, Campisi J, Schilling B (2020). "A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development". PLOS Biology . 18 (1): e3000599. doi: 10.1371/journal.pbio.3000599 . PMC 6964821 . PMID 31945054.
- 1 2 Maciel-Barón LA, Morales-Rosales SL, Aquino-Cruz AA, Königsberg M (2016). "Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli". AGE . 38 (1): 26. doi:10.1007/s11357-016-9886-1. PMC 5005892 . PMID 26867806.
- ↑ Partridge L, Fuentealba M, Kennedy BK (2020). "The quest to slow ageing through drug discovery" (PDF). Nature Reviews Drug Discovery . 19 (8): 513–532. doi:10.1038/s41573-020-0067-7. PMID 32467649. S2CID 218912510.
- 1 2 Chambers ES, Akbar AN (2020). "Can blocking inflammation enhance immunity during aging?". The Journal of Allergy and Clinical Immunology . 145 (5): 1323–1331. doi:10.1016/j.jaci.2020.03.016. PMID 32386656.
- ↑ Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E (2019). "From discoveries in ageing research to therapeutics for healthy ageing". Nature . 571 (7764): 183–192. Bibcode:2019Natur.571..183C. doi: 10.1038/s41586-019-1365-2 . PMC 7205183 . PMID 31292558.
- ↑ Sundeep Khosla S, Farr JN, Tchkonia T, Kirkland JL (2020). "The role of cellular senescence in ageing and endocrine diseasee". Nature Reviews Endocrinology . 16 (5): 263–275. doi:10.1038/s41574-020-0335-y. PMC 7227781 . PMID 32161396.
- ↑ Paez-Ribes M, González-Gualda E, Doherty GJ, Muñoz-Espín D (2019). "Targeting senescent cells in translational medicine". EMBO Molecular Medicine . 11 (12): e10234. doi:10.15252/emmm.201810234. PMC 6895604 . PMID 31746100.
- ↑ Kirkland JL, Tchkonia T (2020). "Senolytic Drugs: From Discovery to Translation". Journal of Internal Medicine . 288 (5): 518–536. doi:10.1111/joim.13141. PMC 7405395 . PMID 32686219.
- 1 2 Booth LN, Brunet A (2016). "The Aging Epigenome". Molecular Cell . 62 (5): 728–744. doi:10.1016/j.molcel.2016.05.013. PMC 4917370 . PMID 27259204.
- 1 2 Ghosh K, Capell BC (2016). "The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging". Journal of Investigative Dermatology . 136 (11): 2133–2139. doi:10.1016/j.jid.2016.06.621. PMC 5526201 . PMID 27543988.
- ↑ Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J (June 2008). "Chemokine signaling via the CXCR2 receptor reinforces senescence". Cell . 133 (6): 1006–18. doi: 10.1016/j.cell.2008.03.038 . PMID 18555777. S2CID 6708172.
- ↑ Yarbro JR, Emmons RS, Pence BD (June 2020). "Macrophage Immunometabolism and Inflammaging: Roles of Mitochondrial Dysfunction, Cellular Senescence, CD38, and NAD". Immunometabolism. 2 (3): e200026. doi:10.20900/immunometab20200026. PMC 7409778 . PMID 32774895.
- 1 2 Kang C, Xu O, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ (2015). "The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4". Science . 349 (6255): aaa5612. doi:10.1126/science.aaa5612. PMC 4942138 . PMID 26404840.
- 1 2 Yessenkyzy A, Saliev T, Zhanaliyeva M, Nurgozhin T (2020). "Polyphenols as Caloric-Restriction Mimetics and Autophagy Inducers in Aging Research". Nutrients . 12 (5): 1344. doi: 10.3390/nu12051344 . PMC 7285205 . PMID 32397145.
- ↑ Rossiello F, Jurk D, Passos JF, di Fagagna F (2022). "Telomere dysfunction in ageing and age-related diseases". Nature Cell Biology . 24 (2): 135–147. doi:10.1038/s41556-022-00842-x. PMC 8985209 . PMID 35165420.
- ↑ Anerillas C, Abdelmohsen K, Gorospe M (2020). "Regulation of senescence traits by MAPKs". GeroScience . 42 (2): 397–408. doi:10.1007/s11357-020-00183-3. PMC 7205942 . PMID 32300964.
- 1 2 Herranz N, Gil J (2018). "Mechanisms and functions of cellular senescence". Journal of Clinical Investigation . 128 (4): 1238–1246. doi:10.1172/JCI95148. PMC 5873888 . PMID 29608137.
- 1 2 Laberge R, Sun Y, Orjalo AV, Patil CK, Campisi J (2015). "MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation". Nature Cell Biology . 17 (8): 1049–1061. doi:10.1038/ncb3195. PMC 4691706 . PMID 26147250.
- ↑ Wang R, Yu Z, Sunchu B, Perez VI (2017). "Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism". Aging Cell . 16 (3): 564–574. doi:10.1111/acel.12587. PMC 5418203 . PMID 28371119.
- ↑ Sharma R, Padwad Y (2019). "In search of nutritional anti-aging targets: TOR inhibitors, SASP modulators, and BCL-2 family suppressors". Nutrition . 65: 33–38. doi:10.1016/j.nut.2019.01.020. PMID 31029919. S2CID 86541289.
- ↑ Wang R, Sunchu B, Perez VI (2017). "Rapamycin and the inhibition of the secretory phenotype". Experimental Gerontology . 94: 89–92. doi:10.1016/j.exger.2017.01.026. PMID 28167236. S2CID 4960885.
- ↑ Weichhart T (2018). "mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review". Gerontology . 84 (2): 127–134. doi:10.1159/000484629. PMC 6089343 . PMID 29190625.
- 1 2 Papadopoli D, Boulay K, Kazak L, Hulea L (2019). "mTOR as a central regulator of lifespan and aging". F1000Research . 8: 998. doi: 10.12688/f1000research.17196.1 . PMC 6611156 . PMID 31316753.
- ↑ Carosi JM, Fourrier C, Bensalem J, Sargeant TJ (2022). "The mTOR-lysosome axis at the centre of ageing". FEBS Open Bio . 12 (4): 739–757. doi:10.1002/2211-5463.13347. PMC 8972043 . PMID 34878722.
- ↑ Paredes S, Angulo-Ibanez M, Tasselli L, Chua KF (2018). "The epigenetic regulator SIRT7 guards against mammalian cellular senescence induced by ribosomal DNA instability". Journal of Biological Chemistry . 293 (28): 11242–11250. doi: 10.1074/jbc.AC118.003325 . PMC 6052228 . PMID 29728458.
- 1 2 Huda N, Liu G, Hong H, Yin X (2019). "Hepatic senescence, the good and the bad". World Journal of Gastroenterology . 25 (34): 5069–5081. doi: 10.3748/wjg.v25.i34.5069 . PMC 6747293 . PMID 31558857.
- ↑ Yang H, Wang H, Ren J, Chen ZJ (2017). "cGAS is essential for cellular senescence". Proceedings of the National Academy of Sciences of the United States of America . 114 (23): E4612–E4620. Bibcode:2017PNAS..114E4612Y. doi: 10.1073/pnas.1705499114 . PMC 5468617 . PMID 28533362.
- 1 2 3 Baechle JJ, Chen N, Winer DA (2023). "Chronic inflammation and the hallmarks of aging". Molecular Metabolism . 74: 101755. doi:10.1016/j.molmet.2023.101755. PMC 10359950 . PMID 37329949.
- 1 2 Liu X, Ding J, Meng L (2018). "Oncogene-induced senescence: a double edged sword in cancer". Acta Pharmacologica Sinica . 39 (10): 1553–1558. doi:10.1038/aps.2017.198. PMC 6289471 . PMID 29620049.
- ↑ Houssaini A, Breau M, Kebe K, Adnot S (2018). "mTOR pathway activation drives lung cell senescence and emphysema". JCI Insight . 3 (3): e93203. doi:10.1172/jci.insight.93203. PMC 5821218 . PMID 29415880.
- ↑ Palmer AK, Gustafson B, Kirkland JL, Smith U (2019). "Cellular senescence: at the nexus between ageing and diabetes". Diabetologia . 62 (10): 1835–1841. doi:10.1007/s00125-019-4934-x. PMC 6731336 . PMID 31451866.
- 1 2 van Deursen JM (2019). "Senolytic therapies for healthy longevity". Science . 364 (6441): 636–637. Bibcode:2019Sci...364..636V. doi:10.1126/science.aaw1299. PMC 6816502 . PMID 31097655.
- ↑ Palmer AK, Kirkland JL (2016). "Aging and adipose tissue: potential interventions for diabetes and regenerative medicine". Experimental Gerontology . 86: 97–105. doi:10.1016/j.exger.2016.02.013. PMC 5001933 . PMID 26924669.
- ↑ Bartleson JM, Radenkovic D, Verdin E (2021). "SARS-CoV-2, COVID-19 and the Ageing Immune System". Nature Aging . 1 (9): 769–782. doi:10.1038/s43587-021-00114-7. PMC 8570568 . PMID 34746804.
- ↑ Chini C, Hogan KA, Warner GM, Tarragó MG, Peclat TR, Tchkonia T, Kirkland JL, Chini E (2019). "The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline". Biochemical and Biophysical Research Communications . 513 (2): 486–493. doi:10.1016/j.bbrc.2019.03.199. PMC 6486859 . PMID 30975470.
- ↑ Eric M. Verdin (2015). "NAD⁺ in aging, metabolism, and neurodegeneration". Science . 350 (6265): 1208–1213. Bibcode:2015Sci...350.1208V. doi:10.1126/science.aac4854. PMID 26785480. S2CID 27313960.
- ↑ Sabbatinelli J, Prattichizzo F, Olivieri F, Giuliani A (2019). "Where Metabolism Meets Senescence: Focus on Endothelial Cells". Frontiers in Physiology . 10: 1523. doi: 10.3389/fphys.2019.01523 . PMC 6930181 . PMID 31920721.
- ↑ Covarrubias AJ, Perrone R, Grozio A, Verdin E (2021). "NAD + metabolism and its roles in cellular processes during ageing". Nature Reviews Molecular Cell Biology . 22 (2): 119–141. doi:10.1038/s41580-020-00313-x. PMC 7963035 . PMID 33353981.
- 1 2 Soto-Gamez A, Quax WJ, Demaria M (2019). "Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions". Journal of Molecular Biology . 431 (15): 2629–2643. doi: 10.1016/j.jmb.2019.05.036 . PMID 31153901.
- 1 2 Prašnikar E, Borišek J, Perdih A (2021). "Senescent cells as promising targets to tackle age-related diseases". Ageing Research Reviews . 66: 101251. doi:10.1016/j.arr.2020.10125. PMID 33385543.
- ↑ Kim YH, Park TJ (2019). "Cellular senescence in cancer". BMB Reports. 52 (1): 42–46. doi:10.5483/BMBRep.2019.52.1.295. PMC 6386235 . PMID 30526772.
- ↑ Lim H, Heo MY, Kim HP (2019). "Flavonoids: Broad Spectrum Agents on Chronic Inflammation". Biomolecules & Therapeutics. 27 (3): 241–253. doi:10.4062/biomolther.2019.034. PMC 6513185 . PMID 31006180.
- ↑ Katlinskaya YV, Carbone CJ, Yu Q, Fuchs SY (2015). "Type 1 interferons contribute to the clearance of senescent cell". Cancer Biology & Therapy. 16 (8): 1214–1219. doi:10.1080/15384047.2015.1056419. PMC 4622626 . PMID 26046815.
- 1 2 Sagiv A, Krizhanovsky V (2013). "Immunosurveillance of senescent cells: the bright side of the senescence program". Biogerontology. 14 (6): 617–628. doi:10.1007/s10522-013-9473-0. PMID 24114507. S2CID 2775067.
- ↑ Thiers, B.H. (January 2008). "Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas". Yearbook of Dermatology and Dermatologic Surgery. 2008: 312–313. doi:10.1016/s0093-3619(08)70921-3. ISSN 0093-3619.
- ↑ Rao, Sonia G.; Jackson, James G. (November 2016). "SASP: Tumor Suppressor or Promoter? Yes!". Trends in Cancer. 2 (11): 676–687. doi: 10.1016/j.trecan.2016.10.001 . ISSN 2405-8033. PMID 28741506.
- ↑ Alexander E, Hildebrand DG, Schulze-Osthoff K, Essmann F (2013). "IκBζ is a regulator of the senescence-associated secretory phenotype in DNA damage- and oncogene-induced senescence". Journal of Cell Science . 126 (Pt 16): 3738–3745. doi: 10.1242/jcs.128835 . PMID 23781024.
- ↑ Yang J, Liu M, Zhang X (2021). "The Paradoxical Role of Cellular Senescence in Cancer". Frontiers in Cell and Developmental Biology . 9: 722205. doi: 10.3389/fcell.2021.722205 . PMC 8388842 . PMID 34458273.
- ↑ Lujambio A (2016). "To clear, or not to clear (senescent cells)? That is the question". BioEssays . 38 (Suppl 1): s56–s64. doi: 10.1002/bies.201670910 . PMID 27417123. S2CID 3785916.
- ↑ Freund A, Orjalo AV, Desprez P, Campisi J (2010). "Inflammatory networks during cellular senescence: causes and consequences". Trends in Molecular Medicine . 16 (5): 238–246. doi:10.1016/j.molmed.2010.03.003. PMC 2879478 . PMID 20444648.
- 1 2 Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dollé ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J (2014). "An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA". Developmental Cell . 31 (6): 722–733. doi:10.1016/j.devcel.2014.11.012. PMC 4349629 . PMID 25499914.
- 1 2 Basisty N, Kale A, Campisi J, Schilling B (2020). "The power of proteomics to monitor senescence-associated secretory phenotypes and beyond: toward clinical applications". Expert Review of Proteomics. 17 (4): 297–308. doi:10.1080/14789450.2020.1766976. PMC 7416420 . PMID 32425074.
- ↑ Muñoz-Espín, Daniel; Serrano, Manuel (July 2014). "Cellular senescence: from physiology to pathology". Nature Reviews Molecular Cell Biology. 15 (7): 482–496. doi:10.1038/nrm3823. ISSN 1471-0080. PMID 24954210. S2CID 20062510.
- ↑ Muñoz-Espín, Daniel; Cañamero, Marta; Maraver, Antonio; Gómez-López, Gonzalo; Contreras, Julio; Murillo-Cuesta, Silvia; Rodríguez-Baeza, Alfonso; Varela-Nieto, Isabel; Ruberte, Jesús; Collado, Manuel; Serrano, Manuel (2013-11-21). "Programmed Cell Senescence during Mammalian Embryonic Development". Cell. 155 (5): 1104–1118. doi: 10.1016/j.cell.2013.10.019 . hdl: 20.500.11940/3668 . ISSN 0092-8674. PMID 24238962.
- ↑ Zhu Y, Liu X, Geng X (2019). "Telomere and its role in the aging pathways: telomere shortening, cell senescence and mitochondria dysfunction". Biogerontology . 20 (1): 1–16. doi:10.1007/s10522-018-9769-1. PMID 30229407.
- ↑ Zhang M, Serna-Salas S, Moshage H (2021). "Hepatic stellate cell senescence in liver fibrosis: Characteristics, mechanisms and perspectives". Mechanisms of Ageing and Development . 199: 111572. doi: 10.1016/j.mad.2021.111572 . ISSN 0047-6374. PMID 34536446. S2CID 237524296.
- ↑ Valentijn FA, Falke LL, Nguyen TQ, Goldschmeding R (2018). "Cellular senescence in the aging and diseased kidney". Journal of Cell Communication and Signaling . 12 (1): 69–82. doi:10.1007/s12079-017-0434-2. PMC 5842195 . PMID 29260442.
- 1 2 Liu GY, Sabatini DM (2020). "mTOR at the nexus of nutrition, growth, ageing and disease". Nature Reviews Molecular Cell Biology . 21 (4): 183–203. doi:10.1038/s41580-019-0199-y. PMC 7102936 . PMID 31937935.
- ↑ Ji S, Xiong M, Sun X (2023). "Cellular rejuvenation: molecular mechanisms and potential therapeutic interventions for diseases". Signal Transduction and Targeted Therapy . 8 (1): 116. doi:10.1038/s41392-023-01343-5. PMC 10015098 . PMID 36918530.
- ↑ Han Y, Ramprasath T, Zou M (2020). "β-hydroxybutyrate and its metabolic effects on age-associated pathology". Experimental & Molecular Medicine . 52 (4): 548–555. doi:10.1038/s12276-020-0415-z. PMC 7210293 . PMID 32269287.
- ↑ Stubbs BJ, Koutnik AP, Volek JS, Newman JC (2021). "From bedside to battlefield: intersection of ketone body mechanisms in geroscience with military resilience". GeroScience . 43 (3): 1071–1081. doi:10.1007/s11357-020-00277-y. PMC 8190215 . PMID 33006708.
- 1 2 Diniz BS, Mulsant BM, Lenze EJ (2022). "Association of Molecular Senescence Markers in Late-Life Depression With Clinical Characteristics and Treatment Outcome". JAMA Network Open . 5 (6): e2219678. doi:10.1001/jamanetworkopen.2022.19678. PMC 9247739 . PMID 35771573.
- ↑ Franceschi C, Campisi J (2014). "Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases". The Journals of Gerontology: Series A . 69 (Supp 1): s4–s9. doi: 10.1093/gerona/glu057 . PMID 24833586.
- ↑ Akbar AN, Gilroy DW (2020). "Aging immunity may exacerbate COVID-19". Science . 369 (6501): 256–257. Bibcode:2020Sci...369..256A. doi: 10.1126/science.abb0762 . PMID 32675364.