Selfish genetic elements are genetic segments that can enhance their own transmission at the expense of other genes in the genome, even if this has no positive or a net negative effect on organismal fitness. Genomes have traditionally been viewed as cohesive units, with genes acting together to improve the fitness of the organism. However, when genes have some control over their own transmission, the rules can change, and so just like all social groups, genomes are vulnerable to selfish behaviour by their parts.

Polyploidy is a condition in which the cells of an organism have more than one pair of (homologous) chromosomes. Most species whose cells have nuclei (eukaryotes) are diploid, meaning they have two complete sets of chromosomes, one from each of two parents; each set contains the same number of chromosomes, and the chromosomes are joined in pairs of homologous chromosomes. However, some organisms are polyploid. Polyploidy is especially common in plants. Most eukaryotes have diploid somatic cells, but produce haploid gametes by meiosis. A monoploid has only one set of chromosomes, and the term is usually only applied to cells or organisms that are normally diploid. Males of bees and other Hymenoptera, for example, are monoploid. Unlike animals, plants and multicellular algae have life cycles with two alternating multicellular generations. The gametophyte generation is haploid, and produces gametes by mitosis; the sporophyte generation is diploid and produces spores by meiosis.

Chromosomal crossover, or crossing over, is the exchange of genetic material during sexual reproduction between two homologous chromosomes' non-sister chromatids that results in recombinant chromosomes. It is one of the final phases of genetic recombination, which occurs in the pachytene stage of prophase I of meiosis during a process called synapsis. Synapsis begins before the synaptonemal complex develops and is not completed until near the end of prophase I. Crossover usually occurs when matching regions on matching chromosomes break and then reconnect to the other chromosome.

Genetic recombination is the exchange of genetic material between different organisms which leads to production of offspring with combinations of traits that differ from those found in either parent. In eukaryotes, genetic recombination during meiosis can lead to a novel set of genetic information that can be further passed on from parents to offspring. Most recombination occurs naturally and can be classified into two types: (1) interchromosomal recombination, occurring through independent assortment of alleles whose loci are on different but homologous chromosomes ; & (2) intrachromosomal recombination, occurring through crossing over.
Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution. It can be defined as any duplication of a region of DNA that contains a gene. Gene duplications can arise as products of several types of errors in DNA replication and repair machinery as well as through fortuitous capture by selfish genetic elements. Common sources of gene duplications include ectopic recombination, retrotransposition event, aneuploidy, polyploidy, and replication slippage.

Comparative genomics is a branch of biological research that examines genome sequences across a spectrum of species, spanning from humans and mice to a diverse array of organisms from bacteria to chimpanzees. This large-scale holistic approach compares two or more genomes to discover the similarities and differences between the genomes and to study the biology of the individual genomes. Comparison of whole genome sequences provides a highly detailed view of how organisms are related to each other at the gene level. By comparing whole genome sequences, researchers gain insights into genetic relationships between organisms and study evolutionary changes. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, Comparative genomics provides a powerful tool for studying evolutionary changes among organisms, helping to identify genes that are conserved or common among species, as well as genes that give unique characteristics of each organism. Moreover, these studies can be performed at different levels of the genomes to obtain multiple perspectives about the organisms.

An inversion is a chromosome rearrangement in which a segment of a chromosome becomes inverted within its original position. An inversion occurs when a chromosome undergoes a two breaks within the chromosomal arm, and the segment between the two breaks inserts itself in the opposite direction in the same chromosome arm. The breakpoints of inversions often happen in regions of repetitive nucleotides, and the regions may be reused in other inversions. Chromosomal segments in inversions can be as small as 1 kilobases or as large as 100 megabases. The number of genes captured by an inversion can range from a handful of genes to hundreds of genes. Inversions can happen either through ectopic recombination between repetitive sequences, or through chromosomal breakage followed by non-homologous end joining.
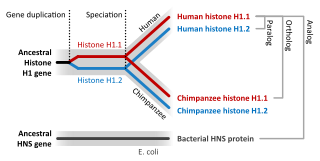
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
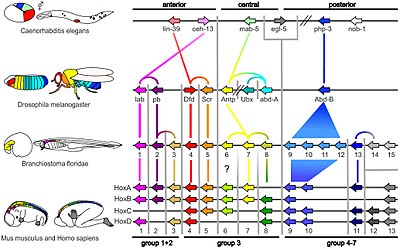
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.

Paleopolyploidy is the result of genome duplications which occurred at least several million years ago (MYA). Such an event could either double the genome of a single species (autopolyploidy) or combine those of two species (allopolyploidy). Because of functional redundancy, genes are rapidly silenced or lost from the duplicated genomes. Most paleopolyploids, through evolutionary time, have lost their polyploid status through a process called diploidization, and are currently considered diploids, e.g., baker's yeast, Arabidopsis thaliana, and perhaps humans.

Copy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals. Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs. Approximately two-thirds of the entire human genome may be composed of repeats and 4.8–9.5% of the human genome can be classified as copy number variations. In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.

Brachypodium distachyon, commonly called purple false brome or stiff brome, is a grass species native to southern Europe, northern Africa and southwestern Asia east to India. It is related to the major cereal grain species wheat, barley, oats, maize, rice, rye, sorghum, and millet. It has many qualities that make it an excellent model organism for functional genomics research in temperate grasses, cereals, and dedicated biofuel crops such as switchgrass. These attributes include small genome diploid accessions, a series of polyploid accessions, a small physical stature, self-fertility, a short lifecycle, simple growth requirements, and an efficient transformation system. The genome of Brachypodium distachyon has been sequenced and published in Nature in 2010.

Masatoshi Nei was a Japanese-born American evolutionary biologist.
In genetics, HAPPY Mapping, first proposed by Paul H. Dear and Peter R. Cook in 1989, is a method used to study the linkage between two or more DNA sequences. According to the Single Molecule Genomics Group, it is "Mapping based on the analysis of approximately HAPloid DNA samples using the PolYmerase chain reaction". In genomics, HAPPY mapping can be applied to assess the synteny and orientation of various DNA sequences across a particular genome - the generation of a "genomic" map.
Population genomics is the large-scale comparison of DNA sequences of populations. Population genomics is a neologism that is associated with population genetics. Population genomics studies genome-wide effects to improve our understanding of microevolution so that we may learn the phylogenetic history and demography of a population.

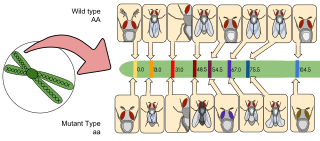
Gene redundancy is the existence of multiple genes in the genome of an organism that perform the same function. Gene redundancy can result from gene duplication. Such duplication events are responsible for many sets of paralogous genes. When an individual gene in such a set is disrupted by mutation or targeted knockout, there can be little effect on phenotype as a result of gene redundancy, whereas the effect is large for the knockout of a gene with only one copy. Gene knockout is a method utilized in some studies aiming to characterize the maintenance and fitness effects functional overlap.

Plant genetics is the study of genes, genetic variation, and heredity specifically in plants. It is generally considered a field of biology and botany, but intersects frequently with many other life sciences and is strongly linked with the study of information systems. Plant genetics is similar in many ways to animal genetics but differs in a few key areas.

Stephen J. O'Brien is an American geneticist. He is known for his research contributions in comparative genomics, virology, genetic epidemiology, mammalian systematics and species conservation. Member of the National Academy of Sciences and a Foreign Member of the Russian Academy of Sciences. Author or co-author of over 850 scientific articles and the editor of fourteen volumes.
The 2000s witnessed an explosion of genome sequencing and mapping in evolutionarily diverse species. While full genome sequencing of mammals is rapidly progressing, the ability to assemble and align orthologous whole chromosomal regions from more than a few species is not yet possible. The intense focus on the building of comparative maps for domestic, laboratory and agricultural (cattle) animals has traditionally been used to understand the underlying basis of disease-related and healthy phenotypes.
Conservation Genomics is the use of genomic study to aide in the preservation and viability of different and diverse organisms and populations. Genomics can be utilized in order to classify or argue diversity, hybridization, and history as well as identity different and similar species. Genomics can evaluate how these measures relate to effective population size as well as other ideas under the umbrella of conservation genetics, and overall biological conservation. Genomic analysis can evaluate the extent to which alleles at certain loci interact with one and other to display nuanced ways which the genome may be intertwined.