Related Research Articles

Oxidative phosphorylation or electron transport-linked phosphorylation or terminal oxidation is the metabolic pathway in which cells use enzymes to oxidize nutrients, thereby releasing chemical energy in order to produce adenosine triphosphate (ATP). In eukaryotes, this takes place inside mitochondria. Almost all aerobic organisms carry out oxidative phosphorylation. This pathway is so pervasive because it releases more energy than alternative fermentation processes such as anaerobic glycolysis.
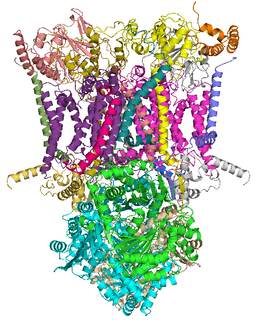
The coenzyme Q : cytochrome c – oxidoreductase, sometimes called the cytochrome bc1 complex, and at other times complex III, is the third complex in the electron transport chain, playing a critical role in biochemical generation of ATP. Complex III is a multisubunit transmembrane protein encoded by both the mitochondrial and the nuclear genomes. Complex III is present in the mitochondria of all animals and all aerobic eukaryotes and the inner membranes of most eubacteria. Mutations in Complex III cause exercise intolerance as well as multisystem disorders. The bc1 complex contains 11 subunits, 3 respiratory subunits, 2 core proteins and 6 low-molecular weight proteins.

Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily.

Cytochrome c oxidase I (COX1) also known as mitochondrially encoded cytochrome c oxidase I (MT-CO1) is a protein that in humans is encoded by the MT-CO1 gene. In other eukaryotes, the gene is called COX1, CO1, or COI. Cytochrome c oxidase I is the main subunit of the cytochrome c oxidase complex. Mutations in MT-CO1 have been associated with Leber's hereditary optic neuropathy (LHON), acquired idiopathic sideroblastic anemia, Complex IV deficiency, colorectal cancer, sensorineural deafness, and recurrent myoglobinuria.

Cytochrome c oxidase subunit 2, also known as cytochrome c oxidase polypeptide II, is a protein that in humans is encoded by the MT-CO2 gene. Cytochrome c oxidase subunit II, abbreviated COXII, COX2, COII, or MT-CO2, is the second subunit of cytochrome c oxidase. It is also one of the three mitochondrial DNA (mtDNA) encoded subunits of respiratory complex IV.
Cytochrome b is a protein that in humans is encoded by the MT-CYB gene. Its gene product is a subunit of the respiratory chain protein ubiquinol–cytochrome c reductase, which consists of the products of one mitochondrially encoded gene, MT-CYB, and ten nuclear genes—UQCRC1, UQCRC2, CYC1, UQCRFS1, UQCRB, "11kDa protein", UQCRH, Rieske protein presequence, "cyt c1 associated protein", and Rieske-associated protein.
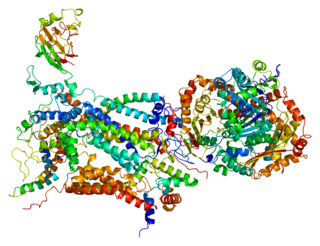
Cytochrome c1, heme protein, mitochondrial (CYC1), also known as UQCR4, MC3DN6, Complex III subunit 4, Cytochrome b-c1 complex subunit 4, or Ubiquinol-cytochrome-c reductase complex cytochrome c1 subunit is a protein that in humans is encoded by the CYC1 gene. CYC1 is a respiratory subunit of Ubiquinol Cytochrome c Reductase, which is located in the inner mitochondrial membrane and is part of the electron transport chain. Mutations in this gene may cause mitochondrial complex III deficiency, nuclear, type 6.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 8, mitochondrial also known as NADH-ubiquinone oxidoreductase 23 kDa subunit, Complex I-23kD (CI-23kD), or TYKY subunit is an enzyme that in humans is encoded by the NDUFS8 gene. The NDUFS8 protein is a subunit of NADH dehydrogenase (ubiquinone) also known as Complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in this gene have been associated with Leigh syndrome.

The human ETFA gene encodes the Electron-transfer-flavoprotein, alpha subunit, also known as ETF-α. Together with Electron-transfer-flavoprotein, beta subunit, encoded by the 'ETFB' gene, it forms the heterodimeric electron transfer flavoprotein (ETF). The native ETF protein contains one molecule of FAD and one molecule of AMP, respectively.

The human ETFB gene encodes the Electron-transfer-flavoprotein, beta subunit, also known as ETF-β. Together with Electron-transfer-flavoprotein, alpha subunit, encoded by the 'ETFA' gene, it forms the heterodimeric Electron transfer flavoprotein (ETF). The native ETF protein contains one molecule of FAD and one molecule of AMP, respectively.

NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial (NDUFS1) is an enzyme that in humans is encoded by the NDUFS1 gene. The encoded protein, NDUFS1, is the largest subunit of complex I, located on the inner mitochondrial membrane, and is important for mitochondrial oxidative phosphorylation. Mutations in this gene are associated with complex I deficiency.

NADH dehydrogenase [ubiquinone] flavoprotein 2, mitochondrial (NDUFV2) is an enzyme that in humans is encoded by the NDUFV2 gene. The encoded protein, NDUFV2, is a subunit of complex I of the mitochondrial respiratory chain, which is located on the inner mitochondrial membrane and involved in oxidative phosphorylation. Mutations in this gene are implicated in Parkinson's disease, bipolar disorder, schizophrenia, and have been found in one case of early onset hypertrophic cardiomyopathy and encephalopathy.

Protoheme IX farnesyltransferase, mitochondrial is an enzyme that in humans is encoded by the COX10 gene. Cytochrome c oxidase (COX), the terminal component of the mitochondrial respiratory chain, catalyzes the electron transfer from reduced cytochrome c to oxygen. This component is a heteromeric complex consisting of 3 catalytic subunits encoded by mitochondrial genes and multiple structural subunits encoded by nuclear genes. The mitochondrially-encoded subunits function in electron transfer, and the nuclear-encoded subunits may function in the regulation and assembly of the complex. This nuclear gene, COX10, encodes heme A: farnesyltransferase, which is not a structural subunit but required for the expression of functional COX and functions in the maturation of the heme A prosthetic group of COX. A gene mutation, which results in the substitution of a lysine for an asparagine (N204K), is identified to be responsible for cytochrome c oxidase deficiency. In addition, this gene is disrupted in patients with CMT1A duplication and with HNPP deletion.

Ubiquinol-cytochrome c reductase binding protein, also known as UQCRB, Complex III subunit 7, QP-C, or Ubiquinol-cytochrome c reductase complex 14 kDa protein is a protein which in humans is encoded by the UQCRB gene. This gene encodes a subunit of the ubiquinol-cytochrome c oxidoreductase complex, which consists of one mitochondrial-encoded and 10 nuclear-encoded subunits. Mutations in this gene are associated with mitochondrial complex III deficiency. Alternatively spliced transcript variants have been found for this gene. Related pseudogenes have been identified on chromosomes 1, 5 and X.

Cytochrome b-c1 complex subunit 2, mitochondrial (UQCRC2), also known as QCR2, UQCR2, or MC3DN5 is a protein that in humans is encoded by the UQCRC2 gene. The product of UQCRC2 is a subunit of the respiratory chain protein Ubiquinol Cytochrome c Reductase, which consists of the products of one mitochondrially encoded gene, MTCYTB and ten nuclear genes: UQCRC1, UQCRC2, Cytochrome c1, UQCRFS1, UQCRB, "11kDa protein", UQCRH, Rieske Protein presequence, "cyt. c1 associated protein", and "Rieske-associated protein." Defects in UQCRC2 are associated with mitochondrial complex III deficiency, nuclear, type 5.

Cytochrome c oxidase assembly protein COX15 homolog (COX15), also known as heme A synthase, is a protein that in humans is encoded by the COX15 gene. This protein localizes to the inner mitochondrial membrane and involved in heme A biosynthesis. COX15 is also part of a three-component mono-oxygenase that catalyses the hydroxylation of the methyl group at position eight of the protoheme molecule. Mutations in this gene has been reported in patients with hypertrophic cardiomyopathy as well as Leigh syndrome, and characterized by delayed onset of symptoms, hypotonia, feeding difficulties, failure to thrive, motor regression, and brain stem signs.

Ubiquinol-cytochrome c reductase, complex III subunit VII, 9.5kDa is a protein that in humans is encoded by the UQCRQ gene. This ubiqinone-binding protein is a subunit of mitochondrial Complex III in the electron transport chain. A mutation in the UQCRQ gene has been shown to cause severe neurological disorders. Infection by Trypanosoma cruzi can cause oxidative modification of this protein in cardiac muscle tissue.
Cytochrome c oxidase assembly factor 5 is a protein that in humans is encoded by the COA5 gene. This gene encodes an ortholog of yeast Pet191, which in yeast is a subunit of a large oligomeric complex associated with the mitochondrial inner membrane, and required for the assembly of the cytochrome c oxidase complex. Mutations in this gene are associated with mitochondrial complex IV deficiency.

Cytochrome c oxidase assembly factor 6 is a protein that in humans is encoded by the COA6 gene. Mitochondrial respiratory chain Complex IV, or cytochrome c oxidase, is the component of the respiratory chain that catalyzes the transfer of electrons from intermembrane space cytochrome c to molecular oxygen in the matrix and as a consequence contributes to the proton gradient involved in mitochondrial ATP synthesis. The COA6 gene encodes an assembly factor for mitochondrial complex IV and is a member of the cytochrome c oxidase subunit 6B family. This protein is located in the intermembrane space, associating with SCO2 and COX2. It stabilizes newly formed COX2 and is part of the mitochondrial copper relay system. Mutations in this gene result in fatal infantile cardioencephalomyopathy.

PET100 homolog is a protein that in humans is encoded by the PET100 gene. Mitochondrial complex IV, or cytochrome c oxidase, is a large transmembrane protein complex that is part of the respiratory electron transport chain of mitochondria. The small protein encoded by the PET100 gene plays a role in the biogenesis of mitochondrial complex IV. This protein localizes to the inner mitochondrial membrane and is exposed to the intermembrane space. Mutations in this gene are associated with mitochondrial complex IV deficiency. This gene has a pseudogene on chromosome 3. Alternative splicing results in multiple transcript variants.
References
- ↑ "CYC1 - Cytochrome c1, heme protein, mitochondrial precursor - Homo sapiens (Human) - CYC1 gene & protein". www.uniprot.org. Retrieved 2016-07-29.
- ↑ Kokhan O, Wraight CA, Tajkhorshid E (October 2010). "The binding interface of cytochrome c and cytochrome c₁ in the bc₁ complex: rationalizing the role of key residues". Biophysical Journal. 99 (8): 2647–56. doi:10.1016/j.bpj.2010.08.042. PMC 2955499 . PMID 20959106.
- ↑ "CYC1 cytochrome c1 [Homo sapiens (human)]". National Center for Biotechnology Information. U.S. National Library of Medicine. Retrieved 2016-07-29.
- ↑ "ortholog_gene_1537[group]". National Center for Biotechnology Information. U.S. National Library of Medicine. Retrieved 2016-07-29.
- ↑ Prince RC, George GN (June 1995). "Cytochrome f revealed". Trends in Biochemical Sciences. 20 (6): 217–8. doi:10.1016/S0968-0004(00)89018-0. PMID 7631417.
- ↑ Online Mendelian Inheritance in Man (OMIM): Cytochrome C!; CYC1 - 123980
- ↑ "OMIM Gene Map". Online Mendelian Inheritance in Man. Johns Hopkins University. Retrieved 2016-07-29.