
Hereditary haemochromatosis type 1 is a genetic disorder characterized by excessive intestinal absorption of dietary iron, resulting in a pathological increase in total body iron stores. Humans, like most animals, have no mechanism to regulate excess iron, simply losing a limited amount through various means like sweating or menstruating.

Thalassemias are inherited blood disorders that result in abnormal hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Symptoms and presentations of thalassemia can change over time. Thalassemia is also known as cooley's anemia or Mediterranean anemia.

Ferritin is a universal intracellular protein that stores iron and releases it in a controlled fashion. The protein is produced by almost all living organisms, including archaea, bacteria, algae, higher plants, and animals. It is the primary intracellular iron-storage protein in both prokaryotes and eukaryotes, keeping iron in a soluble and non-toxic form. In humans, it acts as a buffer against iron deficiency and iron overload.
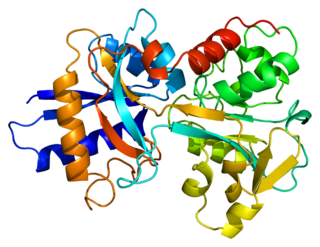
Transferrins are glycoproteins found in vertebrates which bind and consequently mediate the transport of iron (Fe) through blood plasma. They are produced in the liver and contain binding sites for two Fe3+ ions. Human transferrin is encoded by the TF gene and produced as a 76 kDa glycoprotein.

Iron overload or haemochromatosis indicates increased total accumulation of iron in the body from any cause and resulting organ damage. The most important causes are hereditary haemochromatosis, a genetic disorder, and transfusional iron overload, which can result from repeated blood transfusions.
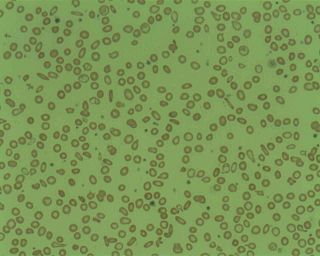
Microcytic anaemia is any of several types of anemia characterized by smaller than normal red blood cells. The normal mean corpuscular volume is approximately 80–100 fL. When the MCV is <80 fL, the red cells are described as microcytic and when >100 fL, macrocytic. The MCV is the average red blood cell size.
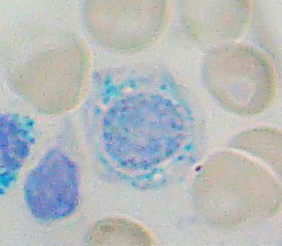
Sideroblastic anemia, or sideroachrestic anemia, is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes). In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome, which can develop into hematological malignancies.

Hemosiderin or haemosiderin is an iron-storage complex that is composed of partially digested ferritin and lysosomes. The breakdown of heme gives rise to biliverdin and iron. The body then traps the released iron and stores it as hemosiderin in tissues. Hemosiderin is also generated from the abnormal metabolic pathway of ferritin.

Total iron-binding capacity (TIBC) or sometimes transferrin iron-binding capacity is a medical laboratory test that measures the blood's capacity to bind iron with transferrin. Transferrin can bind two atoms of ferric iron (Fe3+) with high affinity. It means that transferrin has the capacity to transport approximately from 1.40 to 1.49 mg of iron per gram of transferrin present in the blood.
Transferrin saturation (TS), measured as a percentage, is a medical laboratory value. It is the value of serum iron divided by the total iron-binding capacity of the available transferrin, the main protein that binds iron in the blood, this value tells a clinician how much serum iron is bound. For instance, a value of 15% means that 15% of iron-binding sites of transferrin are being occupied by iron. The three results are usually reported together. A low transferrin saturation is a common indicator of iron deficiency anemia whereas a high transferrin saturation may indicate iron overload or hemochromatosis. Transferrin saturation is also called transferrin saturation index (TSI) or transferrin saturation percentage (TS%)

Human iron metabolism is the set of chemical reactions that maintain human homeostasis of iron at the systemic and cellular level. Iron is both necessary to the body and potentially toxic. Controlling iron levels in the body is a critically important part of many aspects of human health and disease. Hematologists have been especially interested in systemic iron metabolism, because iron is essential for red blood cells, where most of the human body's iron is contained. Understanding iron metabolism is also important for understanding diseases of iron overload, such as hereditary hemochromatosis, and iron deficiency, such as iron-deficiency anemia.
African iron overload is an iron overload disorder first observed among people of African descent in Southern Africa and Central Africa. It is now recognized to actually be two disorders with different causes, possibly compounding each other:
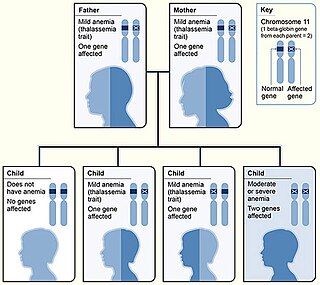
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.

Atransferrinemia is an autosomal recessive metabolic disorder in which there is an absence of transferrin, a plasma protein that transports iron through the blood. Atransferrinemia is characterized by anemia and hemosiderosis in the heart and liver. The iron damage to the heart can lead to heart failure. The anemia is typically microcytic and hypochromic. Atransferrinemia was first described in 1961 and is extremely rare, with only ten documented cases worldwide.
Transfusional hemosiderosis is the accumulation of iron in the body due to frequent blood transfusions. Iron accumulates in the liver and heart, but also endocrine organs. Frequent blood transfusions may be given to many patients, such as those with thalassemia, sickle cell disease, leukemia, aplastic anemia, or myelodysplastic syndrome, among others. It is diagnosed with a blood transferrin test and a liver biopsy. It is treated with venipuncture, erythrocytapheresis, and iron chelation therapy.
Juvenile hemochromatosis, also known as hemochromatosis type 2, is a rare form of hereditary hemochromatosis, which emerges in young individuals, typically between 15 and 30 years of age, but occasionally later. It is characterized by an inability to control how much iron is absorbed by the body, in turn leading to iron overload, where excess iron accumulates in many areas of the body and causes damage to the places it accumulates.

Congenital dyserythropoietic anemia (CDA) is a rare blood disorder, similar to the thalassemias. CDA is one of many types of anemia, characterized by ineffective erythropoiesis, and resulting from a decrease in the number of red blood cells (RBCs) in the body and a less than normal quantity of hemoglobin in the blood. CDA may be transmitted by both parents autosomal recessively or dominantly.
Treatment of the inherited blood disorder thalassemia depends upon the level of severity. For mild forms of the condition, advice and counseling are often all that are necessary. For more severe forms, treatment may consist in blood transfusion; chelation therapy to reverse iron overload, using drugs such as deferoxamine, deferiprone, or deferasirox; medication with the antioxidant indicaxanthin to prevent the breakdown of hemoglobin; or a bone marrow transplant using material from a compatible donor, or from the patient's mother. Removal of the spleen (splenectomy) could theoretically help to reduce the need for blood transfusions in people with thalassaemia major or intermedia but there is currently no reliable evidence from clinical trials about its effects. Population screening has had some success as a preventive measure.
Hemochromatosis type 4 is a hereditary iron overload disorder that affects ferroportin, an iron transport protein needed to export iron from cells into circulation. Although the disease is rare, it is found throughout the world and affects people from various ethnic groups. While the majority of individuals with type 4 hemochromatosis have a relatively mild form of the disease, some affected individuals have a more severe form. As the disease progresses, iron may accumulate in the tissues of affected individuals over time, potentially resulting in organ damage.

Transfusion-dependent anemia is a form of anemia characterized by the need for continuous blood transfusion. It is a condition that results from various diseases, and is associated with decreased survival rates. Regular transfusion is required to reduce the symptoms of anemia by increasing functional red blood cells and hemoglobin count. Symptoms may vary based on the severity of the condition and the most common symptom is fatigue. Various diseases can lead to transfusion-dependent anemia, most notably myelodysplastic syndromes (MDS) and thalassemia. Due to the number of diseases that can cause transfusion-dependent anemia, diagnosing it is more complicated. Transfusion dependence occurs when an average of more than 2 units of blood transfused every 28 days is required over a period of at least 3 months. Myelodysplastic syndromes is often only diagnosed when patients become anemic, and transfusion-dependent thalassemia is diagnosed based on gene mutations. Screening for heterozygosity in the thalassemia gene is an option for early detection.
8. The Guide for the Management of Transfusion Dependent Thalassaemia (TDT) 3rd edition, editors Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V, published and issued by the Thalassaemia International Federation (TIF Publication No23, 2017)


