
Hereditary haemochromatosis type 1 is a genetic disorder characterized by excessive intestinal absorption of dietary iron, resulting in a pathological increase in total body iron stores. Humans, like most animals, have no mechanism to regulate excess iron, simply losing a limited amount through various means like sweating or menstruating.

Ferritin is a universal intracellular protein that stores iron and releases it in a controlled fashion. The protein is produced by almost all living organisms, including archaea, bacteria, algae, higher plants, and animals. It is the primary intracellular iron-storage protein in both prokaryotes and eukaryotes, keeping iron in a soluble and non-toxic form. In humans, it acts as a buffer against iron deficiency and iron overload.
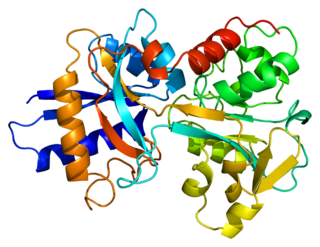
Transferrins are glycoproteins found in vertebrates which bind and consequently mediate the transport of iron (Fe) through blood plasma. They are produced in the liver and contain binding sites for two Fe3+ ions. Human transferrin is encoded by the TF gene and produced as a 76 kDa glycoprotein.

Total iron-binding capacity (TIBC) or sometimes transferrin iron-binding capacity is a medical laboratory test that measures the blood's capacity to bind iron with transferrin. Transferrin can bind two atoms of ferric iron (Fe3+) with high affinity. It means that transferrin has the capacity to transport approximately from 1.40 to 1.49 mg of iron per gram of transferrin present in the blood.
Transferrin saturation (TS), measured as a percentage, is a medical laboratory value. It is the value of serum iron divided by the total iron-binding capacity of the available transferrin, the main protein that binds iron in the blood, this value tells a clinician how much serum iron is bound. For instance, a value of 15% means that 15% of iron-binding sites of transferrin are being occupied by iron. The three results are usually reported together. A low transferrin saturation is a common indicator of iron deficiency anemia whereas a high transferrin saturation may indicate iron overload or hemochromatosis. Transferrin saturation is also called transferrin saturation index (TSI) or transferrin saturation percentage (TS%)

Human iron metabolism is the set of chemical reactions that maintain human homeostasis of iron at the systemic and cellular level. Iron is both necessary to the body and potentially toxic. Controlling iron levels in the body is a critically important part of many aspects of human health and disease. Hematologists have been especially interested in systemic iron metabolism, because iron is essential for red blood cells, where most of the human body's iron is contained. Understanding iron metabolism is also important for understanding diseases of iron overload, such as hereditary hemochromatosis, and iron deficiency, such as iron-deficiency anemia.

Hepcidin is a protein that in humans is encoded by the HAMP gene. Hepcidin is a key regulator of the entry of iron into the circulation in mammals.

Ferroportin-1, also known as solute carrier family 40 member 1 (SLC40A1) or iron-regulated transporter 1 (IREG1), is a protein that in humans is encoded by the SLC40A1 gene. Ferroportin is a transmembrane protein that transports iron from the inside of a cell to the outside of the cell. Ferroportin is the only known iron exporter.

Aceruloplasminemia is a rare autosomal recessive disorder in which the liver can not synthesize the protein ceruloplasmin properly, which is needed to transport copper around the blood. Copper deficiency in the brain results in neurological problems that generally appear in adulthood and worsen over time. .
African iron overload is an iron overload disorder first observed among people of African descent in Southern Africa and Central Africa. It is now recognized to actually be two disorders with different causes, possibly compounding each other:

Human homeostatic iron regulator protein, also known as the HFE protein, is a transmembrane protein that in humans is encoded by the HFE gene. The HFE gene is located on short arm of chromosome 6 at location 6p22.2

Atransferrinemia is an autosomal recessive metabolic disorder in which there is an absence of transferrin, a plasma protein that transports iron through the blood. Atransferrinemia is characterized by anemia and hemosiderosis in the heart and liver. The iron damage to the heart can lead to heart failure. The anemia is typically microcytic and hypochromic. Atransferrinemia was first described in 1961 and is extremely rare, with only ten documented cases worldwide.
Transfusional hemosiderosis is the accumulation of iron in the body due to frequent blood transfusions. Iron accumulates in the liver and heart, but also endocrine organs. Frequent blood transfusions may be given to many patients, such as those with thalassemia, sickle cell disease, leukemia, aplastic anemia, or myelodysplastic syndrome, among others. It is diagnosed with a blood transferrin test and a liver biopsy. It is treated with venipuncture, erythrocytapheresis, and iron chelation therapy.

Iron is an important biological element. It is used in both the ubiquitous iron-sulfur proteins and in vertebrates it is used in hemoglobin which is essential for blood and oxygen transport.

Transferrin receptor 2 (TfR2) is a protein that in humans is encoded by the TFR2 gene. This protein is involved in the uptake of transferrin-bound iron into cells by endocytosis, although its role is minor compared to transferrin receptor 1.
Juvenile hemochromatosis, also known as hemochromatosis type 2, is a rare form of hereditary hemochromatosis, which emerges in young individuals, typically between 15 and 30 years of age, but occasionally later. It is characterized by an inability to control how much iron is absorbed by the body, in turn leading to iron overload, where excess iron accumulates in many areas of the body and causes damage to the places it accumulates.

Hemosiderosis is a form of iron overload disorder resulting in the accumulation of hemosiderin.
Haemochromatosis type 3 is a type of iron overload disorder associated with deficiencies in transferrin receptor 2. It exhibits an autosomal recessive inheritance pattern. The first confirmed case was diagnosed in 1865 by French doctor Trousseau. Later in 1889, the German doctor von Recklinghausen indicated that the liver contains iron, and due to bleeding being considered to be the cause, he called the pigment "Haemochromatosis." In 1935, English doctor Sheldon's groundbreaking book titled, Haemochromatosis, reviewed 311 patient case reports and presented the idea that haemochromatosis was a congenital metabolic disorder. Hereditary haemochromatosis is a congenital disorder which affects the regulation of iron metabolism thus causing increased gut absorption of iron and a gradual build-up of pathologic iron deposits in the liver and other internal organs, joint capsules and the skin. The iron overload could potentially cause serious disease from the age of 40–50 years. In the final stages of the disease, the major symptoms include liver cirrhosis, diabetes and bronze-colored skin. There are four types of hereditary hemochromatosis which are classified depending on the age of onset and other factors such as genetic cause and mode of inheritance.
Hemochromatosis type 4 is a hereditary iron overload disorder that affects ferroportin, an iron transport protein needed to export iron from cells into circulation. Although the disease is rare, it is found throughout the world and affects people from various ethnic groups. While the majority of individuals with type 4 hemochromatosis have a relatively mild form of the disease, some affected individuals have a more severe form. As the disease progresses, iron may accumulate in the tissues of affected individuals over time, potentially resulting in organ damage.
The HFE H63D is a single-nucleotide polymorphism in the HFE gene, which results in the substitution of a histidine for an aspartic acid at amino acid position 63 of the HFE protein (p.His63Asp). HFE participates in the regulation of iron absorption.



