
Familial Mediterranean fever (FMF) is a hereditary inflammatory disorder. FMF is an autoinflammatory disease caused by mutations in Mediterranean fever gene, which encodes a 781–amino acid protein called pyrin. While all ethnic groups are susceptible to FMF, it usually occurs in people of Mediterranean origin—including Sephardic Jews, Mizrahi Jews, Ashkenazi Jews, Assyrians, Armenians, Azerbaijanis, Druze, Levantines, Kurds, Greeks, Turks and Italians.

Hives, also known as urticaria, is a kind of skin rash with red, raised, itchy bumps. Hives may burn or sting. The patches of rash may appear on different body parts, with variable duration from minutes to days, and does not leave any long-lasting skin change. Fewer than 5% of cases last for more than six weeks. The condition frequently recurs.

POEMS syndrome is a rare paraneoplastic syndrome caused by a clone of aberrant plasma cells. The name POEMS is an acronym for some of the disease's major signs and symptoms, as is PEP.

Cold urticaria is a disorder in which large red welts called hives (urticaria) form on the skin after exposure to a cold stimulus. The hives are usually itchy and often the hands and feet will become itchy and swollen as well. Hives vary in size from about 7 mm in diameter to as big as about 27 mm or larger.

Anakinra, sold under the brand name Kineret, is a biopharmaceutical medication used to treat rheumatoid arthritis, cryopyrin-associated periodic syndromes, familial Mediterranean fever, and Still's disease. It is a slightly modified recombinant version of the human interleukin 1 receptor antagonist protein. It is marketed by Swedish Orphan Biovitrum. Anakinra is administered by subcutaneous injection.

Monoclonal gammopathy of undetermined significance (MGUS) is a plasma cell dyscrasia in which plasma cells or other types of antibody-producing cells secrete a myeloma protein, i.e. an abnormal antibody, into the blood; this abnormal protein is usually found during standard laboratory blood or urine tests. MGUS resembles multiple myeloma and similar diseases, but the levels of antibodies are lower, the number of plasma cells in the bone marrow is lower, and it rarely has symptoms or major problems. However, since MGUS can lead to multiple myeloma, which develops at the rate of about 1.5% a year, or other symptomatic conditions, yearly monitoring is recommended.

Pyoderma gangrenosum is a rare, inflammatory skin disease where painful pustules or nodules become ulcers that progressively grow. Pyoderma gangrenosum is not infectious.

Sweet syndrome (SS), or acute febrile neutrophilic dermatosis, is a skin disease characterized by the sudden onset of fever, an elevated white blood cell count, and tender, red, well-demarcated papules and plaques that show dense infiltrates by neutrophil granulocytes on histologic examination.

Muckle–Wells syndrome (MWS) is a rare autosomal dominant disease which causes sensorineural deafness and recurrent hives, and can lead to amyloidosis. Individuals with MWS often have episodic fever, chills, and joint pain. As a result, MWS is considered a type of periodic fever syndrome. MWS is caused by a defect in the CIAS1 gene which creates the protein cryopyrin. MWS is closely related to two other syndromes, familial cold urticaria and neonatal onset multisystem inflammatory disease—in fact, all three are related to mutations in the same gene and subsumed under the term cryopyrin-associated periodic syndromes (CAPS).
Periodic fever syndromes are a set of disorders characterized by recurrent episodes of systemic and organ-specific inflammation. Unlike autoimmune disorders such as systemic lupus erythematosus, in which the disease is caused by abnormalities of the adaptive immune system, people with autoinflammatory diseases do not produce autoantibodies or antigen-specific T or B cells. Instead, the autoinflammatory diseases are characterized by errors in the innate immune system.
Neonatal-onset multisystem inflammatory disease is a rare genetic periodic fever syndrome which causes uncontrolled inflammation in multiple parts of the body starting in the newborn period. Symptoms include skin rashes, severe arthritis, and chronic meningitis leading to neurologic damage. It is one of the cryopyrin-associated periodic syndromes.

Mevalonate kinase deficiency (MKD) is an autosomal recessive metabolic disorder that disrupts the biosynthesis of cholesterol and isoprenoids. It is a very rare genetic disease.
Adult-onset Still's disease (AOSD) is a form of Still's disease, a rare systemic autoinflammatory disease characterized by the classic triad of fevers, joint pain, and a distinctive salmon-colored bumpy rash. The disease is considered a diagnosis of exclusion. Levels of the iron-binding protein ferritin may be extremely elevated with this disorder. AOSD may present in a similar manner to other inflammatory diseases and to autoimmune diseases, which must be ruled out before making the diagnosis.
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare condition (1:1,000,000), in which the bones have lesions, inflammation, and pain. It is called multifocal because it can appear in different parts of the body, primarily bones, and osteomyelitis because it is very similar to that disease, although CRMO appears to be without any infection.
In hematology, plasma cell dyscrasias are a spectrum of progressively more severe monoclonal gammopathies in which a clone or multiple clones of pre-malignant or malignant plasma cells over-produce and secrete into the blood stream a myeloma protein, i.e. an abnormal monoclonal antibody or portion thereof. The exception to this rule is the disorder termed non-secretory multiple myeloma; this disorder is a form of plasma cell dyscrasia in which no myeloma protein is detected in serum or urine of individuals who have clear evidence of an increase in clonal bone marrow plasma cells and/or evidence of clonal plasma cell-mediated tissue injury. Here, a clone of plasma cells refers to group of plasma cells that are abnormal in that they have an identical genetic identity and therefore are descendants of a single genetically distinct ancestor cell.
PAPA syndrome is a rare genetic disorder characterised by its effects on skin and joints. The acronym PAPA stands for pyogenic arthritis, pyoderma gangrenosum and acne.
Cryopyrin-associated periodic syndrome (CAPS) is a group of rare, heterogeneous autoinflammatory disease characterized by interleukin 1β-mediated systemic inflammation and clinical symptoms involving skin, joints, central nervous system, and eyes. It encompasses a spectrum of three clinically overlapping autoinflammatory syndromes including familial cold autoinflammatory syndrome, the Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease that were originally thought to be distinct entities, but in fact share a single genetic mutation and pathogenic pathway, and keratoendotheliitis fugax hereditaria in which the autoinflammatory symptoms affect only the anterior segment of the eye.
Autoinflammatory diseases (AIDs) are a group of rare disorders caused by dysfunction of the innate immune system. These responses are characterized by periodic or chronic systemic inflammation, usually without the involvement of adaptive immunity.
Crystal-storing histiocytosis is a form of histiocytosis that mostly occurs in people with monoclonal gammopathies. Histiocytosis is an excessive number of histiocytes. In the vast majority of crystal-storing histiocytosis cases, immunoglobulins accumulate within the cytoplasm of histiocytes; in rare cases clofazimine, cystine, silica, or Charcot–Leyden crystals may be found in the histiocytes instead. Non-immunoglobulin crystal-storing histiocytosis is mostly associated with non-malignant disorders, such as chronic inflammation or autoimmune abnormality conditions such as rheumatoid arthritis, Crohn's disease, or Helicobacter pylori gastritis. It may be a localised or generalised disease. Examples of locations where histiocytosis may occur include the lungs, pleura, stomach, kidney, bone marrow, thyroid, thymus, and parotid gland. The disease is described as generalised if two or more unrelated sites are involved.
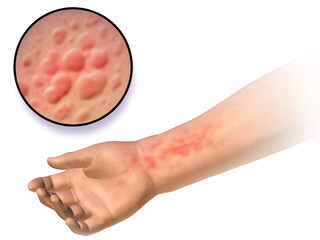
Autoimmune urticaria, also known as chronic autoimmune urticaria, is a type of chronic urticaria characterized by the presence of autoantibodies in the patient's immune system that target the body's own mast cells, leading to episodes of hives (urticaria). This immunologically distinct type of urticaria is considered autoimmune because the immune system, which normally protects the body from foreign organisms, mistakenly attacks the body's own cells, causing inflammation and other symptoms.