
The N-methyl-D-aspartatereceptor (also known as the NMDA receptor or NMDAR), is a glutamate receptor and predominantly Ca2+ ion channel found in neurons. The NMDA receptor is one of three types of ionotropic glutamate receptors, the other two being AMPA and kainate receptors. Depending on its subunit composition, its ligands are glutamate and glycine (or D-serine). However, the binding of the ligands is typically not sufficient to open the channel as it may be blocked by Mg2+ ions which are only removed when the neuron is sufficiently depolarized. Thus, the channel acts as a "coincidence detector" and only once both of these conditions are met, the channel opens and it allows positively charged ions (cations) to flow through the cell membrane. The NMDA receptor is thought to be very important for controlling synaptic plasticity and mediating learning and memory functions.
A muscarinic acetylcholine receptor agonist, also simply known as a muscarinic agonist or as a muscarinic agent, is an agent that activates the activity of the muscarinic acetylcholine receptor. The muscarinic receptor has different subtypes, labelled M1-M5, allowing for further differentiation.
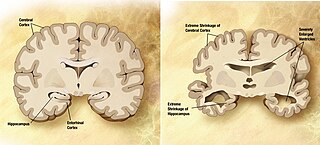
A neurodegenerative disease is caused by the progressive loss of neurons, in the process known as neurodegeneration. Neuronal damage may also ultimately result in their death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

Sphingosine-1-phosphate receptor 5 also known as S1PR5 is a human gene which encodes a G protein-coupled receptor which binds the lipid signaling molecule sphingosine 1-phosphate (S1P). Hence this receptor is also known as S1P5.
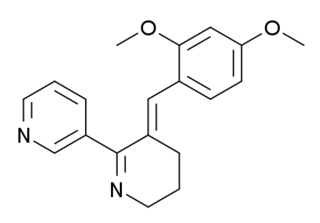
GTS-21 is a drug that has been shown to enhance memory and cognitive function. It has been studied for its potential therapeutic uses, particularly in the treatment of neurodegenerative diseases and psychiatric disorders.
An H3 receptor antagonist is a type of antihistaminic drug used to block the action of histamine at H3 receptors.

Pridopidine is an orally administrated small molecule investigational drug. Pridopidine is a selective and potent Sigma-1 Receptor agonist. It is being developed by Prilenia Therapeutics and is currently in late-stage clinical development for Huntington's disease (HD) and amyotrophic lateral sclerosis (ALS).
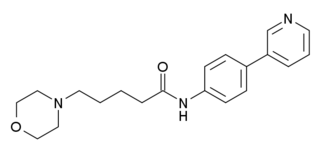
WAY-317538 (SEN-12333) is a drug that acts as a potent and selective full agonist for the α7 subtype of neural nicotinic acetylcholine receptors. It was not the most potent compound in the series, but was selected for further development on the basis of its high selectivity over related receptors, ease of synthesis, and good in vivo properties including high oral bioavailability and good brain penetration. It has nootropic and neuroprotective effects in animal studies, and is being investigated as a potential treatment for neurodegenerative and neurocognitive conditions including Alzheimer's disease and schizophrenia.
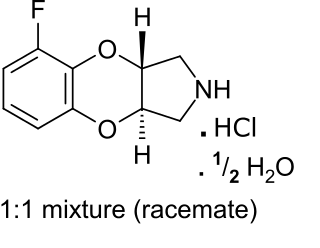
Fluparoxan is a potent α2-adrenergic receptor antagonist with excellent selectivity for this receptor over the α1-adrenergic receptor (2,630-fold), and is the only well-studied α2-adrenergic receptor antagonist in its structural family which does not antagonize any variant of the imidazoline receptor. It was shown to possess central α2-adrenoceptor antagonist activity after oral doses in man and was patented as an antidepressant by Glaxo in the early 1980s, but its development was discontinued when the compound failed to show a clear clinical advantage over existing therapies.

The sphingosine-1-phosphate receptors are a class of G protein-coupled receptors that are targets of the lipid signalling molecule Sphingosine-1-phosphate (S1P). They are divided into five subtypes: S1PR1, S1PR2, S1PR3, S1PR4 and S1PR5.

Neurodegenerative diseases can disrupt the normal human homeostasis and result in abnormal estrogen levels. For example, neurodegenerative diseases can cause different physiological effects in males and females. In particular, estrogen studies have revealed complex interactions with neurodegenerative diseases. Estrogen was initially proposed to be a possible treatment for certain types of neurodegenerative diseases but a plethora of harmful side effects such as increased susceptibility to breast cancer and coronary heart disease overshadowed any beneficial outcomes. On the other hand, Estrogen Replacement Therapy has shown some positive effects with postmenopausal women. Estrogen and estrogen-like molecules form a large family of potentially beneficial alternatives that can have dramatic effects on human homeostasis and disease. Subsequently, large-scale efforts were initiated to screen for useful estrogen family molecules. Furthermore, scientists discovered new ways to synthesize estrogen-like compounds that can avoid many side effects. These are called nonsteroidal estrogens, which come from natural or synthetic products including plants, fungi, and chemicals.
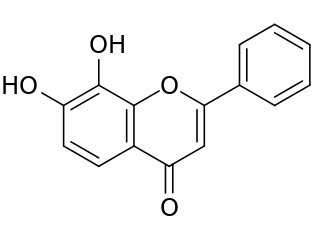
Tropoflavin, also known as 7,8-dihydroxyflavone (DHF), is a naturally occurring flavone found in Godmania aesculifolia, Tridax procumbens, and primula tree leaves. It has been found to act as a potent and selective small-molecule agonist of the tropomyosin receptor kinase B (TrkB), the main signaling receptor of the neurotrophin brain-derived neurotrophic factor (BDNF). Tropoflavin is both orally bioavailable and able to penetrate the blood–brain barrier. A prodrug of tropoflavin with greatly improved potency and pharmacokinetics, R13, is under development for the treatment of Alzheimer's disease.

Ozanimod, sold under the brand name Zeposia, is an immunomodulatory medication for the treatment of relapsing multiple sclerosis and ulcerative colitis. It acts as a sphingosine-1-phosphate receptor (S1PR) agonist, sequestering lymphocytes to peripheral lymphoid organs and away from their sites of chronic inflammation.

Siponimod, sold under the brand name Mayzent, is a selective sphingosine-1-phosphate receptor modulator for oral use that is used for multiple sclerosis (MS). It is intended for once-daily oral administration.

R7 is a small-molecule flavonoid and orally active, potent, and selective agonist of the tropomyosin receptor kinase B (TrkB) – the main signaling receptor for the neurotrophin brain-derived neurotrophic factor (BDNF) – which is under development for the treatment of Alzheimer's disease. It is a structural modification and prodrug of tropoflavin (7,8-DHF) with improved potency and pharmacokinetics, namely oral bioavailability and duration.

Sphingosine-1-phosphate receptor modulators are a class of drugs that interact with S1P receptors, a family of G protein-coupled receptors involved in various physiological processes, particularly in the immune and nervous systems. These modulators have gained significant attention due to their ability to alter lymphocyte trafficking and potentially provide therapeutic benefits in autoimmune diseases, particularly multiple sclerosis (MS). The most well-known compound in this class is fingolimod (FTY720), which was the first oral disease-modifying therapy approved for the treatment of relapsing-remitting MS.
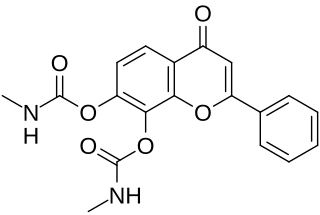
R13 is a small-molecule flavonoid and orally active, potent, and selective agonist of the tropomyosin receptor kinase B (TrkB) – the main signaling receptor for the neurotrophin brain-derived neurotrophic factor (BDNF) – which is under development for the potential treatment of Alzheimer's disease. It is a structural modification and prodrug of tropoflavin (7,8-DHF) with improved potency and pharmacokinetics, namely oral bioavailability and duration. The compound is a replacement for the earlier tropoflavin prodrug R7 and has similar properties to it. It was developed because while R7 displayed a good drug profile in animal studies, it showed almost no conversion into tropoflavin in human liver microsomes. In contrast to R7, R13 is readily hydrolyzed into tropoflavin in human liver microsomes.

Willardiine (correctly spelled with two successive i's) or (S)-1-(2-amino-2-carboxyethyl)pyrimidine-2,4-dione is a chemical compound that occurs naturally in the seeds of Mariosousa willardiana and Acacia sensu lato. The seedlings of these plants contain enzymes capable of complex chemical substitutions that result in the formation of free amino acids (See:#Synthesis). Willardiine is frequently studied for its function in higher level plants. Additionally, many derivates of willardiine are researched for their potential in pharmaceutical development. Willardiine was first discovered in 1959 by R. Gmelin, when he isolated several free, non-protein amino acids from Acacia willardiana (another name for Mariosousa willardiana) when he was studying how these families of plants synthesize uracilyalanines. A related compound, Isowillardiine, was concurrently isolated by a different group, and it was discovered that the two compounds had different structural and functional properties. Subsequent research on willardiine has focused on the functional significance of different substitutions at the nitrogen group and the development of analogs of willardiine with different pharmacokinetic properties. In general, Willardiine is the one of the first compounds studied in which slight changes to molecular structure result in compounds with significantly different pharmacokinetic properties.
Edward Roberts FRSC., is a British-born American scientist with expertise in biochemistry and synthetic organic chemistry. He is recognized for his significant contributions to medicinal chemistry, the design and discovery of new medicines in the development of novel therapeutics.

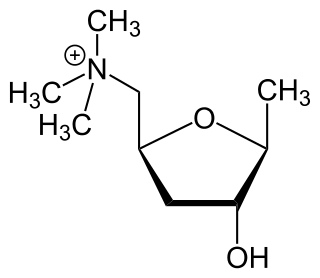
Itameline is a non-selective muscarinic acetylcholine receptor agonist which was under development for the treatment of Alzheimer's disease and memory disorders but was never marketed. It has been referred to as a "nootropic".

