Amino acids are organic compounds that contain both amino and carboxylic acid functional groups. Although over 500 amino acids exist in nature, by far the most important are the 22 α-amino acids incorporated into proteins. Only these 22 appear in the genetic code of life.

Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.

A protein kinase is a kinase which selectively modifies other proteins by covalently adding phosphates to them (phosphorylation) as opposed to kinases which modify lipids, carbohydrates, or other molecules. Phosphorylation usually results in a functional change of the target protein (substrate) by changing enzyme activity, cellular location, or association with other proteins. The human genome contains about 500 protein kinase genes and they constitute about 2% of all human genes. There are two main types of protein kinase. The great majority are serine/threonine kinases, which phosphorylate the hydroxyl groups of serines and threonines in their targets. Most of the others are tyrosine kinases, although additional types exist. Protein kinases are also found in bacteria and plants. Up to 30% of all human proteins may be modified by kinase activity, and kinases are known to regulate the majority of cellular pathways, especially those involved in signal transduction.

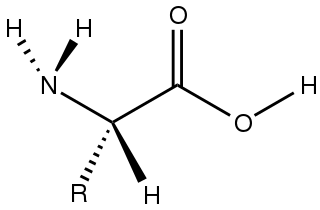
Alanine (symbol Ala or A), or α-alanine, is an α-amino acid that is used in the biosynthesis of proteins. It contains an amine group and a carboxylic acid group, both attached to the central carbon atom which also carries a methyl group side chain. Consequently it is classified as a nonpolar, aliphatic α-amino acid. Under biological conditions, it exists in its zwitterionic form with its amine group protonated (as −NH3+) and its carboxyl group deprotonated (as −CO2−). It is non-essential to humans as it can be synthesized metabolically and does not need to be present in the diet. It is encoded by all codons starting with GC (GCU, GCC, GCA, and GCG).

In molecular biology, post-translational modification (PTM) is the covalent process of changing proteins following protein biosynthesis. PTMs may involve enzymes or occur spontaneously. Proteins are created by ribosomes, which translate mRNA into polypeptide chains, which may then change to form the mature protein product. PTMs are important components in cell signalling, as for example when prohormones are converted to hormones.
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional mutating changes to the DNA sequence of a gene and any gene products. Also called site-specific mutagenesis or oligonucleotide-directed mutagenesis, it is used for investigating the structure and biological activity of DNA, RNA, and protein molecules, and for protein engineering.
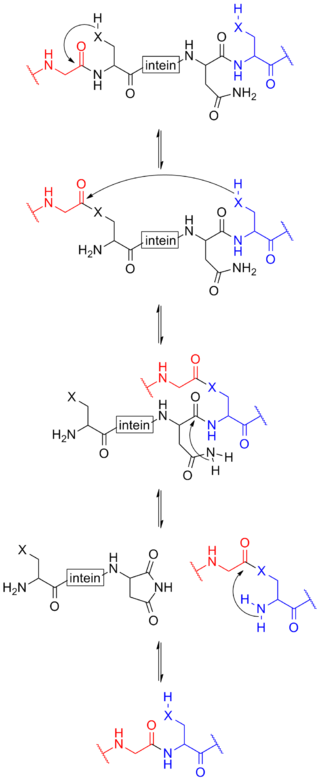
Protein splicing is an intramolecular reaction of a particular protein in which an internal protein segment is removed from a precursor protein with a ligation of C-terminal and N-terminal external proteins on both sides. The splicing junction of the precursor protein is mainly a cysteine or a serine, which are amino acids containing a nucleophilic side chain. The protein splicing reactions which are known now do not require exogenous cofactors or energy sources such as adenosine triphosphate (ATP) or guanosine triphosphate (GTP). Normally, splicing is associated only with pre-mRNA splicing. This precursor protein contains three segments—an N-extein followed by the intein followed by a C-extein. After splicing has taken place, the resulting protein contains the N-extein linked to the C-extein; this splicing product is also termed an extein.

In immunology, epitope mapping is the process of experimentally identifying the binding site, or epitope, of an antibody on its target antigen. Identification and characterization of antibody binding sites aid in the discovery and development of new therapeutics, vaccines, and diagnostics. Epitope characterization can also help elucidate the binding mechanism of an antibody and can strengthen intellectual property (patent) protection. Experimental epitope mapping data can be incorporated into robust algorithms to facilitate in silico prediction of B-cell epitopes based on sequence and/or structural data.
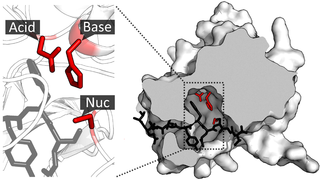
A catalytic triad is a set of three coordinated amino acids that can be found in the active site of some enzymes. Catalytic triads are most commonly found in hydrolase and transferase enzymes. An acid-base-nucleophile triad is a common motif for generating a nucleophilic residue for covalent catalysis. The residues form a charge-relay network to polarise and activate the nucleophile, which attacks the substrate, forming a covalent intermediate which is then hydrolysed to release the product and regenerate free enzyme. The nucleophile is most commonly a serine or cysteine amino acid, but occasionally threonine or even selenocysteine. The 3D structure of the enzyme brings together the triad residues in a precise orientation, even though they may be far apart in the sequence.

The epithelial sodium channel(ENaC), (also known as amiloride-sensitive sodium channel) is a membrane-bound ion channel that is selectively permeable to sodium ions (Na+). It is assembled as a heterotrimer composed of three homologous subunits α or δ, β, and γ, These subunits are encoded by four genes: SCNN1A, SCNN1B, SCNN1G, and SCNN1D. The ENaC is involved primarily in the reabsorption of sodium ions at the collecting ducts of the kidney's nephrons. In addition to being implicated in diseases where fluid balance across epithelial membranes is perturbed, including pulmonary edema, cystic fibrosis, COPD and COVID-19, proteolyzed forms of ENaC function as the human salt taste receptor.
The omega loop is a non-regular protein structural motif, consisting of a loop of six or more amino acid residues and any amino acid sequence. The defining characteristic is that residues that make up the beginning and end of the loop are close together in space with no intervening lengths of regular secondary structural motifs. It is named after its shape, which resembles the upper-case Greek letter Omega (Ω).
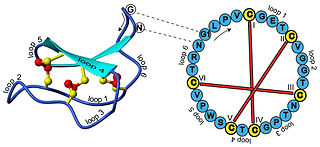
In biochemistry, cyclotides are small, disulfide-rich peptides isolated from plants. Typically containing 28-37 amino acids, they are characterized by their head-to-tail cyclised peptide backbone and the interlocking arrangement of their three disulfide bonds. These combined features have been termed the cyclic cystine knot (CCK) motif. To date, over 100 cyclotides have been isolated and characterized from species of the families Rubiaceae, Violaceae, and Cucurbitaceae. Cyclotides have also been identified in agriculturally important families such as the Fabaceae and Poaceae.

Immunoglobulin heavy constant alpha 1 is a immunoglobulin gene with symbol IGHA1. It encodes a constant (C) segment of Immunoglobulin A heavy chain. Immunoglobulin A is an antibody that plays a critical role in immune function in the mucous membranes. IgA shows the same typical structure of other antibody classes, with two heavy chains and two light chains, and four distinct domains: one variable region, and three variable regions. As a major class of immunoglobulin in body secretions, IgA plays a role in defending against infection, as well as preventing the access of foreign antigens to the immunologic system.

Protein phosphorylation is a reversible post-translational modification of proteins in which an amino acid residue is phosphorylated by a protein kinase by the addition of a covalently bound phosphate group. Phosphorylation alters the structural conformation of a protein, causing it to become activated, deactivated, or otherwise modifying its function. Approximately 13,000 human proteins have sites that are phosphorylated.

Acid-sensing ion channel 3 (ASIC3) also known as amiloride-sensitive cation channel 3 (ACCN3) or testis sodium channel 1 (TNaC1) is a protein that in humans is encoded by the ASIC3 gene. The ASIC3 gene is one of the five paralogous genes that encode proteins that form trimeric acid-sensing ion channels (ASICs) in mammals. The cDNA of this gene was first cloned in 1998. The ASIC genes have splicing variants that encode different proteins that are called isoforms.

Interferon-alpha/beta receptor alpha chain is a protein that in humans is encoded by the IFNAR1 gene.
Hydrophobicity scales are values that define the relative hydrophobicity or hydrophilicity of amino acid residues. The more positive the value, the more hydrophobic are the amino acids located in that region of the protein. These scales are commonly used to predict the transmembrane alpha-helices of membrane proteins. When consecutively measuring amino acids of a protein, changes in value indicate attraction of specific protein regions towards the hydrophobic region inside lipid bilayer.
FoldX is a protein design algorithm that uses an empirical force field. It can determine the energetic effect of point mutations as well as the interaction energy of protein complexes. FoldX can mutate protein and DNA side chains using a probability-based rotamer library, while exploring alternative conformations of the surrounding side chains.
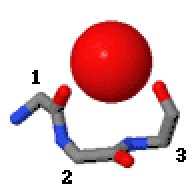
In the area of protein structural motifs, niches are three or four amino acid residue features in which main-chain CO groups are bridged by positively charged or δ+ groups. The δ+ groups include groups with two hydrogen bond donor atoms such as NH2 groups and water molecules. In typical proteins, 7% of amino acid residues belong to niches bound to a δ+ group, while another 7% have the conformation but no single cationic bridging group is detected.
Ribosomally synthesized and post-translationally modified peptides (RiPPs), also known as ribosomal natural products, are a diverse class of natural products of ribosomal origin. Consisting of more than 20 sub-classes, RiPPs are produced by a variety of organisms, including prokaryotes, eukaryotes, and archaea, and they possess a wide range of biological functions.
