
In nuclear physics, beta decay (β-decay) is a type of radioactive decay in which an atomic nucleus emits a beta particle, transforming into an isobar of that nuclide. For example, beta decay of a neutron transforms it into a proton by the emission of an electron accompanied by an antineutrino; or, conversely a proton is converted into a neutron by the emission of a positron with a neutrino in so-called positron emission. Neither the beta particle nor its associated (anti-)neutrino exist within the nucleus prior to beta decay, but are created in the decay process. By this process, unstable atoms obtain a more stable ratio of protons to neutrons. The probability of a nuclide decaying due to beta and other forms of decay is determined by its nuclear binding energy. The binding energies of all existing nuclides form what is called the nuclear band or valley of stability. For either electron or positron emission to be energetically possible, the energy release or Q value must be positive.
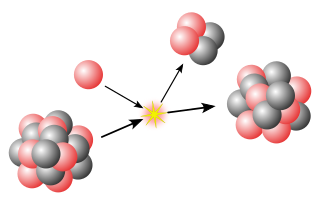
Internal conversion is an atomic decay process where an excited nucleus interacts electromagnetically with one of the orbital electrons of an atom. This causes the electron to be emitted (ejected) from the atom. Thus, in internal conversion, a high-energy electron is emitted from the excited atom, but not from the nucleus. For this reason, the high-speed electrons resulting from internal conversion are not called beta particles, since the latter come from beta decay, where they are newly created in the nuclear decay process.
In chemistry, molecular orbital theory is a method for describing the electronic structure of molecules using quantum mechanics. It was proposed early in the 20th century.
Scintillation is the physical process where a material, called a scintillator, emits UV or visible light under excitation from high energy photons or energetic particles. See scintillator and scintillation counter for practical applications.

In theoretical astrophysics, there can be a sphere of ionized hydrogen around a young star of the spectral classes O or B. The theory was derived by Bengt Strömgren in 1937 and later named Strömgren sphere after him. The Rosette Nebula is the most prominent example of this type of emission nebula from the H II-regions.
A transition metal carbene complex is an organometallic compound featuring a divalent organic ligand. The divalent organic ligand coordinated to the metal center is called a carbene. Carbene complexes for almost all transition metals have been reported. Many methods for synthesizing them and reactions utilizing them have been reported. The term carbene ligand is a formalism since many are not derived from carbenes and almost none exhibit the reactivity characteristic of carbenes. Described often as M=CR2, they represent a class of organic ligands intermediate between alkyls (−CR3) and carbynes (≡CR). They feature in some catalytic reactions, especially alkene metathesis, and are of value in the preparation of some fine chemicals.
Modern valence bond theory is the application of valence bond theory (VBT) with computer programs that are competitive in accuracy and economy with programs for the Hartree–Fock or post-Hartree-Fock methods. The latter methods dominated quantum chemistry from the advent of digital computers because they were easier to program. The early popularity of valence bond methods thus declined. It is only recently that the programming of valence bond methods has improved. These developments are due to and described by Gerratt, Cooper, Karadakov and Raimondi (1997); Li and McWeeny (2002); Joop H. van Lenthe and co-workers (2002); Song, Mo, Zhang and Wu (2005); and Shaik and Hiberty (2004)
In quantum chemistry, a configuration state function (CSF), is a symmetry-adapted linear combination of Slater determinants. A CSF must not be confused with a configuration. In general, one configuration gives rise to several CSFs; all have the same total quantum numbers for spin and spatial parts but differ in their intermediate couplings.
The Hückel method or Hückel molecular orbital theory, proposed by Erich Hückel in 1930, is a simple method for calculating molecular orbitals as linear combinations of atomic orbitals. The theory predicts the molecular orbitals for π-electrons in π-delocalized molecules, such as ethylene, benzene, butadiene, and pyridine. It provides the theoretical basis for Hückel's rule that cyclic, planar molecules or ions with π-electrons are aromatic. It was later extended to conjugated molecules such as pyridine, pyrrole and furan that contain atoms other than carbon and hydrogen (heteroatoms). A more dramatic extension of the method to include σ-electrons, known as the extended Hückel method (EHM), was developed by Roald Hoffmann. The extended Hückel method gives some degree of quantitative accuracy for organic molecules in general and was used to provide computational justification for the Woodward–Hoffmann rules. To distinguish the original approach from Hoffmann's extension, the Hückel method is also known as the simple Hückel method (SHM).
Unrestricted Hartree–Fock (UHF) theory is the most common molecular orbital method for open shell molecules where the number of electrons of each spin are not equal. While restricted Hartree–Fock theory uses a single molecular orbital twice, one multiplied by the α spin function and the other multiplied by the β spin function in the Slater determinant, unrestricted Hartree–Fock theory uses different molecular orbitals for the α and β electrons. This has been called a different orbitals for different spins (DODS) method. The result is a pair of coupled Roothaan equations, known as the Pople–Nesbet–Berthier equations.
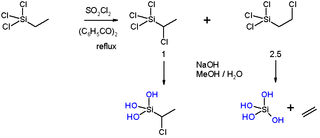
The beta-silicon effect also called silicon hyperconjugation in organosilicon chemistry is a special type of hyperconjugation that describes the stabilizing influence of a silicon atom on the development of positive charge at a carbon atom one position removed (β) from the silicon atom. The C-Si σ orbital is said to partially overlap with the σ* anti-bonding orbital of the C-leaving group, lowering the energy of the transition state leading to the formation of a carbocation. A prerequisite for the hyperconjugation to occur is an antiperiplanar relationship between the Si group and the leaving group. This allows for the maximum overlap between the C-Si σ orbital and the σ* anti-bonding orbital of the leaving group. Silicon hyperconjugation explains specific observations regarding chemical kinetics and stereochemistry of organic reactions with reactants containing silicon.

In organic chemistry, Möbius aromaticity is a special type of aromaticity believed to exist in a number of organic molecules. In terms of molecular orbital theory these compounds have in common a monocyclic array of molecular orbitals in which there is an odd number of out-of-phase overlaps, the opposite pattern compared to the aromatic character to Hückel systems. The nodal plane of the orbitals, viewed as a ribbon, is a Möbius strip, rather than a cylinder, hence the name. The pattern of orbital energies is given by a rotated Frost circle (with the edge of the polygon on the bottom instead of a vertex), so systems with 4n electrons are aromatic, while those with 4n + 2 electrons are anti-aromatic/non-aromatic. Due to incrementally twisted nature of the orbitals of a Möbius aromatic system, stable Möbius aromatic molecules need to contain at least 8 electrons, although 4 electron Möbius aromatic transition states are well known in the context of the Dewar-Zimmerman framework for pericyclic reactions. Möbius molecular systems were considered in 1964 by Edgar Heilbronner by application of the Hückel method, but the first such isolable compound was not synthesized until 2003 by the group of Rainer Herges. However, the fleeting trans-C9H9+ cation, one conformation of which is shown on the right, was proposed to be a Möbius aromatic reactive intermediate in 1998 based on computational and experimental evidence.
STO-nG basis sets are minimal basis sets, where primitive Gaussian orbitals are fitted to a single Slater-type orbital (STO). originally took the values 2 – 6. They were first proposed by John Pople. A minimum basis set is where only sufficient orbitals are used to contain all the electrons in the neutral atom. Thus for the hydrogen atom, only a single 1s orbital is needed, while for a carbon atom, 1s, 2s and three 2p orbitals are needed. The core and valence orbitals are represented by the same number of primitive Gaussian functions . For example, an STO-3G basis set for the 1s, 2s and 2p orbital of the carbon atom are all linear combination of 3 primitive Gaussian functions. For example, a STO-3G s orbital is given by:
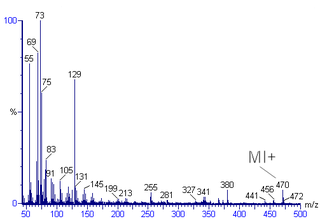
Mass spectral interpretation is the method employed to identify the chemical formula, characteristic fragment patterns and possible fragment ions from the mass spectra. Mass spectra is a plot of relative abundance against mass-to-charge ratio. It is commonly used for the identification of organic compounds from electron ionization mass spectrometry. Organic chemists obtain mass spectra of chemical compounds as part of structure elucidation and the analysis is part of many organic chemistry curricula.
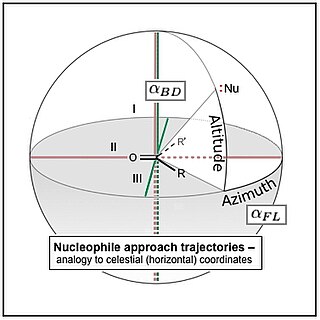
The Flippin–Lodge angle is one of two angles used by organic and biological chemists studying the relationship between a molecule's chemical structure and ways that it reacts, for reactions involving "attack" of an electron-rich reacting species, the nucleophile, on an electron-poor reacting species, the electrophile. Specifically, the angles—the Bürgi–Dunitz, , and the Flippin–Lodge, —describe the "trajectory" or "angle of attack" of the nucleophile as it approaches the electrophile, in particular when the latter is planar in shape. This is called a nucleophilic addition reaction and it plays a central role in the biological chemistry taking place in many biosyntheses in nature, and is a central "tool" in the reaction toolkit of modern organic chemistry, e.g., to construct new molecules such as pharmaceuticals. Theory and use of these angles falls into the areas of synthetic and physical organic chemistry, which deals with chemical structure and reaction mechanism, and within a sub-specialty called structure correlation.
Difluorocarbene is the chemical compound with formula CF2. It has a short half-life, 0.5 and 20 ms, in solution and in the gas phase, respectively. Although highly reactive, difluorocarbene is an intermediate in the production of tetrafluoroethylene, which is produced on an industrial scale as the precursor to Teflon (PTFE).
In Hückel theory, a Dewar reactivity number, also known as Dewar number, is a measure of the reactivity in aromatic systems. It is used to quantify the difference in energy between the π-system of the original molecule and the intermediate having the incoming electrophile or nucleophile attached. It can be used to study important transformations such as the nitration of conjugated systems from a theoretical perspective.
The Edwards equation in organic chemistry is a two-parameter equation for correlating nucleophilic reactivity, as defined by relative rate constants, with the basicity of the nucleophile and its polarizability. This equation was first developed by John O. Edwards in 1954 and later revised based on additional work in 1956.
A Fischer carbene is a type of transition metal carbene complex, which is an organometallic compound containing a divalent organic ligand. In a Fischer carbene, the carbene ligand is a σ-donor π-acceptor ligand. Because π-backdonation from the metal centre is generally weak, the carbene carbon is electrophilic.
Generally speaking, second-row elements such as silicon (Si) are known to stabilize α-carbanions with greater effectiveness than a first-row element, which also means Si could destabilize the α-carbocations. This effect is known as silicon alpha effect. Another term that always associates with silicon alpha effect is the so-called silicon beta effect, which means Si at the β position could support formation of carbocations.

