Related Research Articles

The alpha helix (α-helix) is a common motif in the secondary structure of proteins and is a right hand-helix conformation in which every backbone N−H group hydrogen bonds to the backbone C=O group of the amino acid located four residues earlier along the protein sequence.

Protein secondary structure is the three dimensional form of local segments of proteins. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
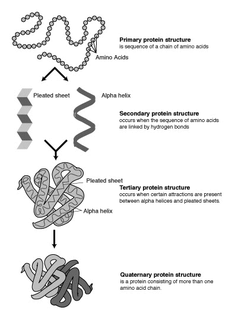
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology. The problem has been solved using artificial intelligence by DeepMind, a London-based AI-company owned by Google, that developed the predictive tool AlphaFold. AlphaFold is a revolutionary artificial intelligence system that has successfully predicted the structures of more than 200 million proteins from approximately 1 million species, covering virtually the entire protein universe known to humans. The structural reservoir containing all predictions has been set up by DeepMind jointly with the European Bioinformatics Institute, an organism within the European Molecular Biology Laboratory (EMBL–EBI) based in Cambridge, UK.
In a chain-like biological molecule, such as a protein or nucleic acid, a structural motif is a common three-dimensional structure that appears in a variety of different, evolutionarily unrelated molecules. A structural motif does not have to be associated with a sequence motif; it can be represented by different and completely unrelated sequences in different proteins or RNA.
A coiled coil is a structural motif in proteins in which 2–7 alpha-helices are coiled together like the strands of a rope. Many coiled coil-type proteins are involved in important biological functions, such as the regulation of gene expression — e.g., transcription factors. Notable examples are the oncoproteins c-Fos and c-Jun, as well as the muscle protein tropomyosin.

A leucine zipper is a common three-dimensional structural motif in proteins. They were first described by Landschulz and collaborators in 1988 when they found that an enhancer binding protein had a very characteristic 30-amino acid segment and the display of these amino acid sequences on an idealized alpha helix revealed a periodic repetition of leucine residues at every seventh position over a distance covering eight helical turns. The polypeptide segments containing these periodic arrays of leucine residues were proposed to exist in an alpha-helical conformation and the leucine side chains from one alpha helix interdigitate with those from the alpha helix of a second polypeptide, facilitating dimerization.
A DNA-binding domain (DBD) is an independently folded protein domain that contains at least one structural motif that recognizes double- or single-stranded DNA. A DBD can recognize a specific DNA sequence or have a general affinity to DNA. Some DNA-binding domains may also include nucleic acids in their folded structure.

A pi helix is a type of secondary structure found in proteins. Discovered by crystallographer Barbara Low in 1952 and once thought to be rare, short π-helices are found in 15% of known protein structures and are believed to be an evolutionary adaptation derived by the insertion of a single amino acid into an α-helix. Because such insertions are highly destabilizing, the formation of π-helices would tend to be selected against unless it provided some functional advantage to the protein. π-helices therefore are typically found near functional sites of proteins.

A 310 helix is a type of secondary structure found in proteins and polypeptides. Of the numerous protein secondary structures present, the 310-helix is the fourth most common type observed; following α-helices, β-sheets and reverse turns. 310-helices constitute nearly 10–15% of all helices in protein secondary structures, and are typically observed as extensions of α-helices found at either their N- or C- termini. Because of the α-helices tendency to consistently fold and unfold, it has been proposed that the 310-helix serves as an intermediary conformation of sorts, and provides insight into the initiation of α-helix folding.
A helix bundle is a small protein fold composed of several alpha helices that are usually nearly parallel or antiparallel to each other.

The tetratricopeptide repeat (TPR) is a structural motif. It consists of a degenerate 34 amino acid tandem repeat identified in a wide variety of proteins. It is found in tandem arrays of 3–16 motifs, which form scaffolds to mediate protein–protein interactions and often the assembly of multiprotein complexes. These alpha-helix pair repeats usually fold together to produce a single, linear solenoid domain called a TPR domain. Proteins with such domains include the anaphase-promoting complex (APC) subunits cdc16, cdc23 and cdc27, the NADPH oxidase subunit p67-phox, hsp90-binding immunophilins, transcription factors, the protein kinase R (PKR), the major receptor for peroxisomal matrix protein import PEX5, protein arginine methyltransferase 9 (PRMT9), and mitochondrial import proteins.
β turns are the most common form of turns—a type of non-regular secondary structure in proteins that cause a change in direction of the polypeptide chain. They are very common motifs in proteins and polypeptides. Each consists of four amino acid residues. They can be defined in two ways:
- By the possession of an intra-main-chain hydrogen bond between the CO of residue i and the NH of residue i+3;
- By having a distance of less than 7Å between the Cα atoms of residues i and i+3.
The Walker A and Walker B motifs are protein sequence motifs, known to have highly conserved three-dimensional structures. These were first reported in ATP-binding proteins by Walker and co-workers in 1982.

Schellman loops are commonly occurring structural features of proteins and polypeptides. Each has six amino acid residues with two specific inter-mainchain hydrogen bonds and a characteristic main chain dihedral angle conformation. The CO group of residue i is hydrogen-bonded to the NH of residue i+5, and the CO group of residue i+1 is hydrogen-bonded to the NH of residue i+4. Residues i+1, i+2, and i+3 have negative φ (phi) angle values and the phi value of residue i+4 is positive. Schellman loops incorporate a three amino acid residue RL nest, in which three mainchain NH groups form a concavity for hydrogen bonding to carbonyl oxygens. About 2.5% of amino acids in proteins belong to Schellman loops. Two websites are available for examining small motifs in proteins, Motivated Proteins: ; or PDBeMotif:.
The Asx turn is a structural feature in proteins and polypeptides. It consists of three amino acid residues in which residue i is an aspartate (Asp) or asparagine (Asn) that forms a hydrogen bond from its sidechain CO group to the mainchain NH group of residue i+2. About 14% of Asx residues present in proteins belong to Asx turns.
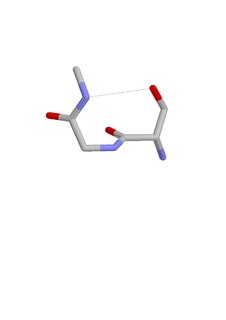
The ST turn is a structural feature in proteins and polypeptides. Each consists of three amino acid residues in which residue i is a serine (S) or threonine (T) that forms a hydrogen bond from its sidechain oxygen group to the mainchain NH group of residue i + 2.
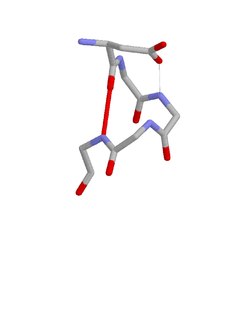
The Asx motif is a commonly occurring feature in proteins and polypeptides. It consists of four or five amino acid residues with either aspartate or asparagine as the first residue. It is defined by two internal hydrogen bonds. One is between the side chain oxygen of residue i and the main chain NH of residue i+2 or i+3; the other is between the main chain oxygen of residue i and the main chain NH of residue i+3 or i+4. Asx motifs occur commonly in proteins and polypeptides.

The ST motif is a commonly occurring feature in proteins and polypeptides. It consists of four or five amino acid residues with either serine or threonine as the first residue. It is defined by two internal hydrogen bonds. One is between the side chain oxygen of residue i and the main chain NH of residue i + 2 or i + 3; the other is between the main chain oxygen of residue i and the main chain NH of residue i + 3 or i + 4. Two websites are available for finding and examining ST motifs in proteins, Motivated Proteins: and PDBeMotif.
The term N cap describes an amino acid in a particular position within a protein or polypeptide. The N cap residue of an alpha helix is the first amino acid residue at the N terminus of the helix. More precisely, it is defined as the first residue (i) whose CO group is hydrogen-bonded to the NH group of residue i+4. Because of this it is sometimes also described as the residue prior to the helix.

Transmembrane epididymal protein 1 is a transmembrane protein encoded by the TEDDM1 gene. TEDDM1 is also commonly known as TMEM45C and encodes 273 amino acids that contains six alpha-helix transmembrane regions. The protein contains a 118 amino acid length family of unknown function. While the exact function of TEDDM1 is not understood, it is predicted to be an integral component of the plasma membrane.
References
- ↑ Richardson, JM; Richardson DC (1988). "Amino acid preferences for specific locations at the ends of alpha-helices". Science. 240 (4859): 1648–1652. Bibcode:1988Sci...240.1648R. doi:10.1126/science.3381086. PMID 3381086. S2CID 38467101.
- ↑ Presta, LG; Rose GD (1988). "Helix Caps". Science. 240 (4859): 1632–1641. Bibcode:1988Sci...240.1632P. doi:10.1126/science.2837824. PMID 2837824.
- ↑ Doig, AJ; MacArthur MW (1997). "Structures of N-termini of helices in proteins". Protein Science. 6 (1): 147–155. doi:10.1002/pro.5560060117. PMC 2143508 . PMID 9007987.
- ↑ Aurora, R; Rose GD (1998). "Helix Capping". Protein Science. 7 (1): 21–38. doi:10.1002/pro.5560070103. PMC 2143812 . PMID 9514257.
- ↑ Gunasekaran, K; Nagarajam HA (1998). "Stereochemical punctuation marks in protein structure". Journal of Molecular Biology. 275 (5): 917–932. doi:10.1006/jmbi.1997.1505. PMID 9480777. S2CID 35919397.
- ↑ Leader, DP; Milner-White EJ (2011). "The structure of the ends of helices in globular proteins". Proteins. 79 (3): 1010–1019. doi:10.1002/prot.22942. PMID 21287629. S2CID 22240314.