Related Research Articles

A catecholamine is a monoamine neurotransmitter, an organic compound that has a catechol and a side-chain amine.
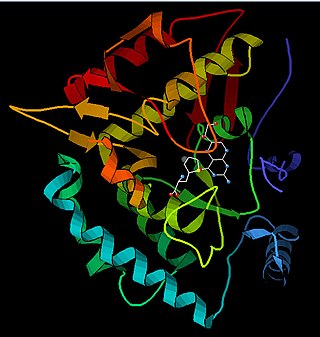
Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.

Tetrahydrobiopterin (BH4, THB), also known as sapropterin (INN), is a cofactor of the three aromatic amino acid hydroxylase enzymes, used in the degradation of amino acid phenylalanine and in the biosynthesis of the neurotransmitters serotonin (5-hydroxytryptamine, 5-HT), melatonin, dopamine, norepinephrine (noradrenaline), epinephrine (adrenaline), and is a cofactor for the production of nitric oxide (NO) by the nitric oxide synthases. Chemically, its structure is that of a (dihydropteridine reductase) reduced pteridine derivative (quinonoid dihydrobiopterin).
The vesicular monoamine transporter (VMAT) is a transport protein integrated into the membranes of synaptic vesicles of presynaptic neurons. It transports monoamine neurotransmitters – such as dopamine, serotonin, norepinephrine, epinephrine, and histamine – into the vesicles, which release the neurotransmitters into synapses, as chemical messages to postsynaptic neurons. VMATs utilize a proton gradient generated by V-ATPases in vesicle membranes to power monoamine import.

Pterin is a heterocyclic compound composed of a pteridine ring system, with a "keto group" and an amino group on positions 4 and 2 respectively. It is structurally related to the parent bicyclic heterocycle called pteridine. Pterins, as a group, are compounds related to pterin with additional substituents. Pterin itself is of no biological significance.

The dopamine transporter (DAT) also is a membrane-spanning protein coded for in the human by the SLC6A3 gene,, that pumps the neurotransmitter dopamine out of the synaptic cleft back into cytosol. In the cytosol, other transporters sequester the dopamine into vesicles for storage and later release. Dopamine reuptake via DAT provides the primary mechanism through which dopamine is cleared from synapses, although there may be an exception in the prefrontal cortex, where evidence points to a possibly larger role of the norepinephrine transporter.

α-Methyl-p-tyrosine (AMPT), or simply α-methyltyrosine, also known in its chiral 2-(S) form as metirosine, is a tyrosine hydroxylase enzyme inhibitor and is therefore a drug involved in inhibiting the catecholamine biosynthetic pathway. AMPT inhibits tyrosine hydroxylase whose enzymatic activity is normally regulated through the phosphorylation of different serine residues in regulatory domain sites. Catecholamine biosynthesis starts with dietary tyrosine, which is hydroxylated by tyrosine hydroxylase and it is hypothesized that AMPT competes with tyrosine at the tyrosine-binding site, causing inhibition of tyrosine hydroxylase.
Tetrahydrobiopterin deficiency (THBD, BH4D) is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained normally through the diet, but can be harmful if excess levels build up, causing intellectual disability and other serious health problems. In healthy individuals, it is metabolised (hydroxylated) into tyrosine, another amino acid, by phenylalanine hydroxylase. However, this enzyme requires tetrahydrobiopterin as a cofactor and thus its deficiency slows phenylalanine metabolism.

6-Pyruvoyltetrahydropterin synthase deficiency is an autosomal recessive disorder that causes malignant hyperphenylalaninemia due to tetrahydrobiopterin deficiency. It is a recessive disorder that is accompanied by hyperphenylalaninemia. Commonly reported symptoms are initial truncal hypotonia, subsequent appendicular hypertonia, bradykinesia, cogwheel rigidity, generalized dystonia, and marked diurnal fluctuation. Other reported clinical features include difficulty in swallowing, oculogyric crises, somnolence, irritability, hyperthermia, and seizures. Chorea, athetosis, hypersalivation, rash with eczema, and sudden death have also been reported. Patients with mild phenotypes may deteriorate if given folate antagonists such as methotrexate, which can interfere with a salvage pathway through which dihydrobiopterin is converted into tetrahydrobiopterin via dihydrofolate reductase. Treatment options include substitution with neurotransmitter precursors, monoamine oxidase inhibitors, and tetrahydrobiopterin. Response to treatment is variable and the long-term and functional outcome is unknown. To provide a basis for improving the understanding of the epidemiology, genotype–phenotype correlation and outcome of these diseases, their impact on the quality of life of patients, and for evaluating diagnostic and therapeutic strategies a patient registry was established by the noncommercial International Working Group on Neurotransmitter Related Disorders (iNTD).

GTP cyclohydrolase I (GTPCH) (EC 3.5.4.16) is a member of the GTP cyclohydrolase family of enzymes. GTPCH is part of the folate and biopterin biosynthesis pathways. It is responsible for the hydrolysis of guanosine triphosphate (GTP) to form 7,8-dihydroneopterin triphosphate (7,8-DHNP-3'-TP, 7,8-NH2-3'-TP).
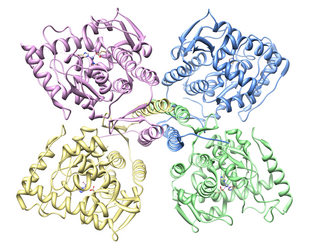
Tyrosine hydroxylase or tyrosine 3-monooxygenase is the enzyme responsible for catalyzing the conversion of the amino acid L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA). It does so using molecular oxygen (O2), as well as iron (Fe2+) and tetrahydrobiopterin as cofactors. L-DOPA is a precursor for dopamine, which, in turn, is a precursor for the important neurotransmitters norepinephrine (noradrenaline) and epinephrine (adrenaline). Tyrosine hydroxylase catalyzes the rate limiting step in this synthesis of catecholamines. In humans, tyrosine hydroxylase is encoded by the TH gene, and the enzyme is present in the central nervous system (CNS), peripheral sympathetic neurons and the adrenal medulla. Tyrosine hydroxylase, phenylalanine hydroxylase and tryptophan hydroxylase together make up the family of aromatic amino acid hydroxylases (AAAHs).

Tryptophan hydroxylase (TPH) is an enzyme (EC 1.14.16.4) involved in the synthesis of the monoamine neurotransmitter serotonin. Tyrosine hydroxylase, phenylalanine hydroxylase, and tryptophan hydroxylase together constitute the family of biopterin-dependent aromatic amino acid hydroxylases. TPH catalyzes the following chemical reaction

Biopterins are pterin derivatives which function as endogenous enzyme cofactors in many species of animals and in some bacteria and fungi. The prototypical compound of the class is biopterin, as shown in the infobox. Biopterins act as cofactors for aromatic amino acid hydroxylases (AAAH), which are involved in synthesizing a number of neurotransmitters including dopamine, norepinephrine, epinepherine, and serotonin, along with several trace amines. Nitric oxide synthesis also uses biopterin derivatives as cofactors. In humans, tetrahydrobiopterin (BH4) is the endogenous cofactor for AAAH enzymes.
Alkylglycerol monooxygenase (AGMO) is an enzyme that catalyzes the hydroxylation of alkylglycerols, a specific subclass of ether lipids. This enzyme was first described in 1964 as a pteridine-dependent ether lipid cleaving enzyme. In 2010 finally, the gene coding for alkylglycerol monooxygenase was discovered as transmembrane protein 195 (TMEM195) on chromosome 7. In analogy to the enzymes phenylalanine hydroxylase, tyrosine hydroxylase, tryptophan hydroxylase and nitric oxide synthase, alkylglycerol monooxygenase critically depends on the cofactor tetrahydrobiopterin and iron.

GTP cyclohydrolase 1 feedback regulatory protein is an enzyme that in humans is encoded by the GCHFR gene.
Dopamine-responsive dystonia (DRD) also known as Segawa syndrome (SS), is a genetic movement disorder which usually manifests itself during early childhood at around ages 5–8 years.

DBH-like monooxygenase protein 1, also known as monooxygenase X, is an enzyme that in humans is encoded by the MOXD1 gene.

Biopterin-dependent aromatic amino acid hydroxylases (AAAH) are a family of aromatic amino acid hydroxylase enzymes which includes phenylalanine 4-hydroxylase, tyrosine 3-hydroxylase, and tryptophan 5-hydroxylase. These enzymes primarily hydroxylate the amino acids L-phenylalanine, L-tyrosine, and L-tryptophan, respectively.
The TH gene codes for the enzyme tyrosine hydroxylase.
Autosomal dominant GTP cyclohydrolase I deficiency (AD-GTPCHD) is a disease caused by dysfunction of GTP cyclohydrolase I, an enzyme that plays an important role in the synthesis of tetrahydrobiopterin, and, as a consequence, of dopamine. This condition is one of the six known causes of tetrahydrobiopterin deficiency and is the most frequently-reported cause of dopa-responsive dystonia.
References
- 1 2 3 Stathakis DG, Burton DY, McIvor WE, Krishnakumar S, Wright TR, O'Donnell JM (September 1999). "The catecholamines up (Catsup) protein of Drosophila melanogaster functions as a negative regulator of tyrosine hydroxylase activity". Genetics. 153 (1): 361–82. PMC 1460756 . PMID 10471719.
- ↑ O’Donnell, Janis M.; Stathakis, Dean G.; Burton, Denise; Chen, Zuomin (2002). Chemistry and Biology of Pteridines and Folates. Springer, Boston, MA. pp. 211–215. doi:10.1007/978-1-4615-0945-5_35. ISBN 9781461353171.
- 1 2 3 4 5 6 7 8 Wang Z, Ferdousy F, Lawal H, Huang Z, Daigle JG, Izevbaye I, Doherty O, Thomas J, Stathakis DG, O'Donnell JM (December 2011). "Catecholamines up integrates dopamine synthesis and synaptic trafficking". Journal of Neurochemistry. 119 (6): 1294–305. doi:10.1111/j.1471-4159.2011.07517.x. PMC 3233821 . PMID 21985068.
- 1 2 3 4 5 6 Groth C, Sasamura T, Khanna MR, Whitley M, Fortini ME (July 2013). "Protein trafficking abnormalities in Drosophila tissues with impaired activity of the ZIP7 zinc transporter Catsup". Development. 140 (14): 3018–27. doi:10.1242/dev.088336. PMC 3699284 . PMID 23785054.
- ↑ Carbone MA, Jordan KW, Lyman RF, Harbison ST, Leips J, Morgan TJ, DeLuca M, Awadalla P, Mackay TF (May 2006). "Phenotypic variation and natural selection at catsup, a pleiotropic quantitative trait gene in Drosophila". Current Biology. 16 (9): 912–9. doi: 10.1016/j.cub.2006.03.051 . PMC 10766118 . PMID 16682353.
- 1 2 3 Hsouna A, Lawal HO, Izevbaye I, Hsu T, O'Donnell JM (August 2007). "Drosophila dopamine synthesis pathway genes regulate tracheal morphogenesis". Developmental Biology. 308 (1): 30–43. doi:10.1016/j.ydbio.2007.04.047. PMC 1995089 . PMID 17585895.
- ↑ Jeong J, Eide DJ (2013). "The SLC39 family of zinc transporters". Molecular Aspects of Medicine. 34 (2–3): 612–9. doi:10.1016/j.mam.2012.05.011. PMC 3602797 . PMID 23506894.