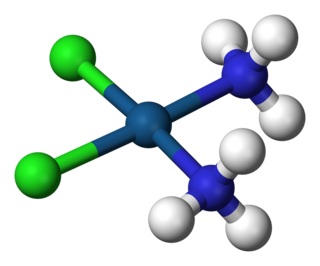
A coordination complex is a chemical compound consisting of a central atom or ion, which is usually metallic and is called the coordination centre, and a surrounding array of bound molecules or ions, that are in turn known as ligands or complexing agents. Many metal-containing compounds, especially those that include transition metals, are coordination complexes.
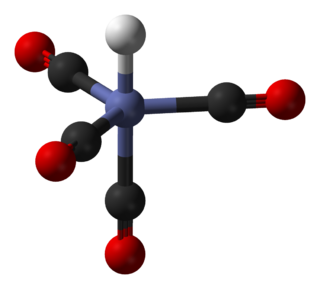
In coordination chemistry, a ligand is an ion or molecule with a functional group that binds to a central metal atom to form a coordination complex. The bonding with the metal generally involves formal donation of one or more of the ligand's electron pairs, often through Lewis bases. The nature of metal–ligand bonding can range from covalent to ionic. Furthermore, the metal–ligand bond order can range from one to three. Ligands are viewed as Lewis bases, although rare cases are known to involve Lewis acidic "ligands".

Organometallic chemistry is the study of organometallic compounds, chemical compounds containing at least one chemical bond between a carbon atom of an organic molecule and a metal, including alkali, alkaline earth, and transition metals, and sometimes broadened to include metalloids like boron, silicon, and selenium, as well. Aside from bonds to organyl fragments or molecules, bonds to 'inorganic' carbon, like carbon monoxide, cyanide, or carbide, are generally considered to be organometallic as well. Some related compounds such as transition metal hydrides and metal phosphine complexes are often included in discussions of organometallic compounds, though strictly speaking, they are not necessarily organometallic. The related but distinct term "metalorganic compound" refers to metal-containing compounds lacking direct metal-carbon bonds but which contain organic ligands. Metal β-diketonates, alkoxides, dialkylamides, and metal phosphine complexes are representative members of this class. The field of organometallic chemistry combines aspects of traditional inorganic and organic chemistry.
A substitution reaction is a chemical reaction during which one functional group in a chemical compound is replaced by another functional group. Substitution reactions are of prime importance in organic chemistry. Substitution reactions in organic chemistry are classified either as electrophilic or nucleophilic depending upon the reagent involved, whether a reactive intermediate involved in the reaction is a carbocation, a carbanion or a free radical, and whether the substrate is aliphatic or aromatic. Detailed understanding of a reaction type helps to predict the product outcome in a reaction. It also is helpful for optimizing a reaction with regard to variables such as temperature and choice of solvent.
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound (also known as organostannanes). A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
In chemistry, π backbonding is a π-bonding interaction between a filled (or half filled) orbital of a transition metal atom and a vacant orbital on an adjacent ion or molecule. In this type of interaction, electrons from the metal are used to bond to the ligand, which dissipates excess negative charge and stabilizes the metal. It is common in transition metals with low oxidation states that have ligands such as carbon monoxide, olefins, or phosphines. The ligands involved in π backbonding can be broken into three groups: carbonyls and nitrogen analogs, alkenes and alkynes, and phosphines. Compounds where π backbonding is prominent include Ni(CO)4, Zeise's salt, and molybdenum and iron dinitrogen complexes.
Reductive elimination is an elementary step in organometallic chemistry in which the oxidation state of the metal center decreases while forming a new covalent bond between two ligands. It is the microscopic reverse of oxidative addition, and is often the product-forming step in many catalytic processes. Since oxidative addition and reductive elimination are reverse reactions, the same mechanisms apply for both processes, and the product equilibrium depends on the thermodynamics of both directions.
Oxidative addition and reductive elimination are two important and related classes of reactions in organometallic chemistry. Oxidative addition is a process that increases both the oxidation state and coordination number of a metal centre. Oxidative addition is often a step in catalytic cycles, in conjunction with its reverse reaction, reductive elimination.

β-Hydride elimination is a reaction in which an alkyl group bonded to a metal centre is converted into the corresponding metal-bonded hydride and an alkene. The alkyl must have hydrogens on the β-carbon. For instance butyl groups can undergo this reaction but methyl groups cannot. The metal complex must have an empty site cis to the alkyl group for this reaction to occur. Moreover, for facile cleavage of the C–H bond, a d electron pair is needed for donation into the σ* orbital of the C–H bond. Thus, d0 metals alkyls are generally more stable to β-hydride elimination than d2 and higher metal alkyls and may form isolable agostic complexes, even if an empty coordination site is available.
In inorganic chemistry, the trans effect is the increased lability of ligands that are trans to certain other ligands, which can thus be regarded as trans-directing ligands. It is attributed to electronic effects and it is most notable in square planar complexes, although it can also be observed for octahedral complexes. The analogous cis effect is most often observed in octahedral transition metal complexes.
In organometallic chemistry, a migratory insertion is a type of reaction wherein two ligands on a metal complex combine. It is a subset of reactions that very closely resembles the insertion reactions, and both are differentiated by the mechanism that leads to the resulting stereochemistry of the products. However, often the two are used interchangeably because the mechanism is sometimes unknown. Therefore, migratory insertion reactions or insertion reactions, for short, are defined not by the mechanism but by the overall regiochemistry wherein one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:

Organorhodium chemistry is the chemistry of organometallic compounds containing a rhodium-carbon chemical bond, and the study of rhodium and rhodium compounds as catalysts in organic reactions.
Marcetta York Darensbourg is an American inorganic chemist. She is a Distinguished Professor of Chemistry at Texas A&M University. Her current work focuses on iron hydrogenases and iron nitrosyl complexes.

A metal-phosphine complex is a coordination complex containing one or more phosphine ligands. Almost always, the phosphine is an organophosphine of the type R3P (R = alkyl, aryl). Metal phosphine complexes are useful in homogeneous catalysis. Prominent examples of metal phosphine complexes include Wilkinson's catalyst (Rh(PPh3)3Cl), Grubbs' catalyst, and tetrakis(triphenylphosphine)palladium(0).
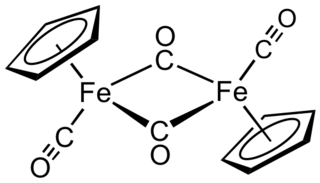
Cyclopentadienyliron dicarbonyl dimer is an organometallic compound with the formula [(η5-C5H5)Fe(CO)2]2, often abbreviated to Cp2Fe2(CO)4, [CpFe(CO)2]2 or even Fp2, with the colloquial name "fip dimer". It is a dark reddish-purple crystalline solid, which is readily soluble in moderately polar organic solvents such as chloroform and pyridine, but less soluble in carbon tetrachloride and carbon disulfide. Cp2Fe2(CO)4 is insoluble in but stable toward water. Cp2Fe2(CO)4 is reasonably stable to storage under air and serves as a convenient starting material for accessing other Fp (CpFe(CO)2) derivatives (described below).
The covalent bond classification (CBC) method, also referred to as LXZ notation, is a way of describing covalent compounds such as organometallic complexes in a way that is not prone to limitations resulting from the definition of oxidation state. Instead of simply assigning a charge to an atom in the molecule, the covalent bond classification method analyzes the nature of the ligands surrounding the atom of interest. According to this method, the interactions that allow for coordination of the ligand can be classified according to whether it donates two, one, or zero electrons. These three classes of ligands are respectively given the symbols L, X, and Z. The method was published by Malcolm L. H. Green in 1995.
In covalent bond classification, a Z-type ligand refers to a ligand that accepts two electrons from the metal center. This is in contrast to X-type ligands, which form a bond with the ligand and metal center each donating one electron, and L-type ligands, which form a bond with the ligand donating two electrons. Typically, these Z-type ligands are Lewis acids, or electron acceptors. They are also known as zero-electron reagents.
In organometallic chemistry, a transition metal alkyne complex is a coordination compound containing one or more alkyne ligands. Such compounds are intermediates in many catalytic reactions that convert alkynes to other organic products, e.g. hydrogenation and trimerization.
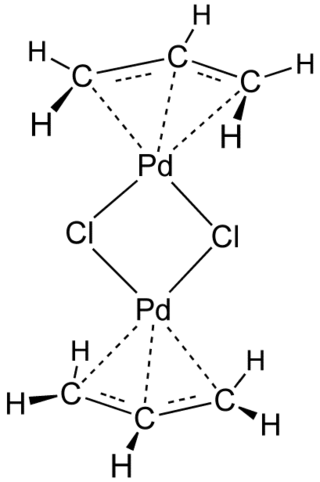
Transition-metal allyl complexes are coordination complexes with allyl and its derivatives as ligands. Allyl is the radical with the connectivity CH2CHCH2, although as a ligand it is usually viewed as an allyl anion CH2=CH−CH2−, which is usually described as two equivalent resonance structures.
In chemistry, transition metal silyl complexes describe coordination complexes in which a transition metal is bonded to an anionic silyl ligand, forming a metal-silicon sigma bond. This class of complexes are numerous and some are technologically significant as intermediates in hydrosilylation. These complexes are a subset of organosilicon compounds.
